Tobacco Product Listing; Form FDA 3742 Listing of Ingredients (Electronic and Paper submissions); Section 904(a)(1)
Tobacco Product Establishment Registration and Submission of Certain Health Information
0650_Guidance_2023
Tobacco Product Listing; Form FDA 3742 Listing of Ingredients (Electronic and Paper submissions); Section 904(a)(1)
OMB: 0910-0650
Registration and Product Listing for Owners and Operators of Domestic Tobacco Product Establishments (Revised*)

Guidance for Industry
Comments may be submitted at any time for Agency consideration. Electronic comments may be submitted to http://www.regulations.gov. Alternatively, submit written comments to the Division of Dockets Management (HFA-305), Food and Drug Administration, 5630 Fishers Lane, Room 1061, Rockville, MD 20852. All comments should be identified with Docket No. FDA-2009-D- 0500.
For questions regarding this guidance, contact the Center for Tobacco Products at 1-877-CTP- 1373 (1-877-287-1373), Monday – Friday, 9 a.m. – 4 p.m. EDT.
Additional copies are available online at http://www.fda.gov/TobaccoProducts/Labeling/RulesRegulationsGuidance/default.htm. You may send an e-mail request to SmallBiz.Tobacco@fda.hhs.gov to receive an electronic copy of this guidance. You may send a request for hard copies to U.S. Food and Drug Administration, Center for Tobacco Products, Attn: Office of Small Business Assistance,
Document Control Center, Bldg. 71, Rm. G335, 10903 New Hampshire Ave., Silver Spring, MD 20993-2000.
U.S. Department of Health and Human Services Food and Drug Administration
Center for Tobacco Products Month 2023
OMB control number: 0910-0650 Current expiration date available at https://www.reginfo.gov
See additional PRA statement in section V of the guidance.
* This is the eighth revision to the first edition of this guidance, which issued in November 2009. Revisions are noted by date at the end of the guidance.
Table of Contents
-
Who Registers Their Establishment and Submits Product Listing Information Under Section 905 of the Act? 5
What Information is Submitted as Part of Establishment Registration and Product Listing Submissions? 7
Registration Information 6
Product Listing Information 7
How to Make Registration and Product Listing Submissions 9
When Must You Register Your Establishment and List Your Products Under
Section 905 of the FD&C Act? 9
APPENDIX A – EXAMPLE PACKAGE LABEL PLAN 14
Registration and Product Listing for Owners and Operators of Domestic Tobacco Product Establishments

Guidance for Industry1

This guidance document is intended to assist owners and operators of domestic tobacco product establishments with the registration and product listing submissions required by section 905 of the Food, Drug, and Cosmetic Act (FD&C Act).2 The guidance document explains, among other things:
The statutory requirement to make establishment registration and product listing submissions;
The definitions of terms used in the statute and this guidance;
Who should make establishment registration and product listing submissions;
What information to include in the submissions;
How to submit the information;
When to submit the information; and
FDA’s compliance policies.
FDA’s guidance documents, including this guidance, do not establish legally enforceable responsibilities. Instead, guidances describe the Agency’s current thinking
on a topic and should be viewed only as recommendations, unless specific regulatory or statutory requirements are cited. The use of the word should in Agency guidances means that something is suggested or recommended, but not required.
The Family Smoking Prevention and Tobacco Control Act (Tobacco Control Act), enacted on June 22, 2009, amends the FD&C Act and provides FDA with the authority to regulate the manufacture, marketing, and distribution of tobacco products to protect the public health (Pub. L. 111– 31, 123 Stat. 1776.) Among other things, the Tobacco Control Act adds section 905 to the FD&C Act (21 U.S.C. 387e), requiring the owners and operators of domestic manufacturing establishments engaged in manufacturing tobacco products to register with FDA and submit product listings.
Cigarettes, cigarette tobacco, roll-your-own tobacco (RYO), and smokeless tobacco were immediately covered by FDA’s tobacco product authorities in chapter IX of the Act, including section 905, when the Tobacco Control Act went into effect. On March 15, 2022, the statutory definition of “tobacco products” was amended to include products “containing nicotine from any source,” and section 901(b) of the Act was amended to make tobacco products containing nicotine not made or derived from tobacco subject to the requirements of chapter IX of the Act, including section 905 of the FD&C Act (Pub. L. 117-103). As for other types of tobacco products, section 901(b) of the FD&C Act (21 U.S.C. 387a(b)) grants FDA authority to deem those products subject to chapter IX of the FD&C Act. Pursuant to that authority, FDA issued a proposed rule seeking to deem all other products that meet the statutory definition of tobacco product set forth in section 201(rr) of the FD&C Act (21 U.S.C. 321(rr)) (except for accessories of those products) (79 FR 23142, April 25, 2014).3 After review and consideration of comments on the proposed rule, the final rule published on May 10, 2016 (81 FR 28974) (the deeming rule) with the effective date of August 8, 2016. As a result, owners and operators of domestic establishments engaged in the manufacture, preparation, compounding, or processing of tobacco products subject to the deeming rule are now required to comply with chapter IX of the FD&C Act, including the establishment registration and product listing requirements in section 905. 4
Section 905(b) of the FD&C Act requires that “every person who owns or operates any establishment in any State engaged in the manufacture, preparation, compounding, or processing of a tobacco product or tobacco products” register with FDA the name, places of business, and all establishments engaged in these activities owned or operated by that person. Following the initial registration, every person must register annually by December 31 of each year.
Section 905(i)(1) of the FD&C Act requires that all registrants “shall, at the time of registration… file with [FDA] a list of all tobacco products which are being manufactured, prepared, compounded, or processed by that person for commercial distribution,” a long with certain accompanying information, including all labeling. In addition, section 905(i)(3) of the FD&C Act requires that certain changes in the product list be submitted biannually.
The failure to register in accordance with section 905 of the FD&C Act, the failure to provide any information required by section 905(i), or the failure to provide a notice required by section 905(i)(3) is a prohibited act under section 301(p) of the FD&C Act (21 U.S.C. 331(p)). In addition, under section 903(a)(6) of the FD&C Act (21 U.S.C. 387c), a tobacco product is dee med misbranded if it was manufactured, prepared, propagated, compounded, or processed in an establishment not duly registered under section 905 or if it was not included in a list required by section 905(i). Violations relating to registration and product listing under section 905 are subject to regulatory and enforcement action, including, but not limited to, seizure and injunction. Although such actions may be authorized, it is the Agency’s practice to give individua ls and firms an opportunity to take voluntary and prompt corrective action before it initiates an enforcement action when doing so is consistent with the FDA’s public protection responsibilities. An advisory action letter, such as a Warning Letter, may be issued by FDA to achieve voluntary compliance.
FDA intends to use the following definitions in implementing the establishment registration and product listing requirements of section 905 of the FD&C Act.
Accessory: The term accessory means any product that is intended or reasonably expected to be used with or for the human consumption of a tobacco product; does not contain tobacco and is not made or derived from tobacco; and meets either of the following:
Is not intended or reasonably expected to affect or alter the performance, composition, constituents, or characteristics of a tobacco product; or
Is intended or reasonably expected to affect or maintain the performance, composition, constituents, or characteristics of a tobacco product but
(i) Solely controls moisture and/or temperature of a stored tobacco product; or
Solely provides an external heat source to initiate but not maintain combustion of a tobacco product.
Certain Tobacco Product Compliance Deadlines Related to the Final Deeming Rule, available at https://www.fda.gov/downloads/TobaccoProducts/Labeling/RulesRegulationsGuidance/UCM557716.pdf.
Commercial distribution: The term commercial distribution means any distribution of a tobacco product to consumers or to another person through sale or otherwise, but does not include internal or interplant transfer of a tobacco product between registered establishments within the same parent, subsidiary, and/or affiliate company, nor does it include providing a tobacco product for product testing where such products are not made available for consumption or resale. For example, the term includes the distribution of a tobacco product as a promotional sample and the delivery of a tobacco product to another manufacturer for further processing via contract without a change in the forma l ownership of the product.
Component or part: The term component or part means any software or assembly of materials intended or reasonably expected:
To alter or affect the tobacco product’s performance, composition, constituents, or characteristics; or
To be used with or for the human consumption of a tobacco product. Component or part excludes anything that is an accessory of a tobacco product.
FDA notes that component and part are separate and distinct terms within chapter IX of the FD&C Act. However, for purposes of this guidance, FDA is using the terms component and part interchangeably and without emphasizing the distinction. FDA may clarify the distinctions between component and part in the future.
Domestic establishment: The term domestic establishment means an establishment in any State or Territory or possession of the United States.
Establishment: The term establishment means a place of business under one ownership at one general physical location. A single building ma y house more than one distinct establishment if the establishments are under separate ownership.
Finished tobacco product: The term finished tobacco product means a tobacco product, including all components and parts, sealed in final packaging intended for consumer use (e.g., filters or filter tubes sold separately to consumers or as part of kits).
Labeling: The term labeling, based on section 201(m) of the FD&C Act, means all labels and other written, printed, or graphic matter (1) upon any tobacco product or any of its containers or wrappers, or (2) accompanying such tobacco product.
Manufacturing: The term manufacturing means the manufacture, preparation, compounding, or processing of a tobacco product, including repackaging or otherwise changing the container, wrapper, or labeling of any tobacco product package (section 905(a)(1) of the FD&C Act). This term includes the activities of reconstituting and blending tobacco leaf; testing for quality control and product release; and applying any chemical, additive, or substance to the tobacco lea f other than potable water in the form of steam or mist. This term excludes the activities of de-stemming, drying, or packing tobacco leaf; mechanically re moving foreign material from tobacco leaves; and humidifying tobacco lea f with nothing other than potable water in the form of steam or mist.
Owner: The term owner means a person, as defined in section 201(e) of the FD&C Act, who has an ownership interest in an establishment.
Operator: The term operator means a person, as defined in section 201(e) of the FD&C Act, who has management authority over an establishment.
Tobacco product: The term tobacco product is defined in section 201(rr) of the FD&C Act,
which states in relevant part:
The term “tobacco product” means any product made or derived from tobacco, or containing nicotine from any source, that is intended for human consumption, including any component, part, or accessory of a tobacco product (except for raw materials other than tobacco used in manufacturing a component, part, or accessory of a tobacco product).
The term “tobacco product” does not mean an article that is a drug under [section 201(g)(1)]; a device under [section 201(h)]; a combination product described in section 503(g) [of the FD&C Act]; or a food under [section 201(f)] if such article contains no nicotine, or no more than trace amounts of naturally occurring nicotine.
Note that this definition includes accessories and components and parts of tobacco products, whether they contain nicotine, or are made or derived from tobacco, and whether they are sold or distributed as finished tobacco products.5
Who Registers their Establishment and Submits Product Listing Information Under Section 905 of the FD&C Act?
Every person who owns or operates any domestic establishment engaged in manufacturing regulated tobacco products must register under section 905(b) of the FD&C Act, and every registrant must file a list of its regulated tobacco products in accordance with section 905(i) of the FD&C Act. An owner or operator ma y authorize a third-party agent to register their establishment and submit product listing information on its behalf. Establishment registration and product listing requirements currently apply only to those persons who own or operate domestic establishments engaged in manufacturing tobacco products; an importer who does not own or operate such an establishment is not subject to the requirements of section 905(b) or section 905(i) of the FD&C Act.
To reduce redundant submissions, FDA strongly encourages that the owner act as the agent for all operators within a given business structure in submitting establishment registration and product listing information. Under this approach, the owner would register all establishments it owns and submit the associated product listing information, and the owner would also register on behalf of all operators with management authority over those establishments. An owner acting as the agent for one or more operators would need to submit all information required of the operator(s) (e.g., the operator name(s) and place(s) of business), but could submit all information for both the owner and the operator(s) in a single registration. If an owner registers/lists all of its establishments using this approach, the operators of those establishments would not need to register. For scenarios in which an owner is also the operator of a given establishment, the owner/operator can register in a single registration.
The following example illustrates the registration and product listing requirements for a complex business structure: Firm A owns establishment X, which is engaged in manufacturing tobacco products. Firm B does not own an establishment engaged in manufacturing tobacco products. Firm A and Firm B are wholly owned subsidiaries of Holding Company C. Holding Company C does not own or operate any establishments engaged in manufacturing tobacco products. In this scenario, Firm A would be required to register establishment X and list products, but neither Firm B nor Holding Company C would be subject to registration or product listing requirements.
FDA’s Compliance Policy for Regulated Tobacco Products
At this time, with respect to all tobacco products, FDA intends to enforce the registration and listing requirements of section 905 with respect to finished tobacco products only (excluding accessories). FDA does not, at this time, intend to enforce these requirements with respect to products that are sold or distributed solely for further manufacturing.
As defined above, the term finished tobacco product means a tobacco product, including all components and parts, sealed in final packaging intended for consumer use (e.g., filters or filter tubes sold separately to consumers or as part of kits).
Components and parts that are sold separately from other tobacco products are also finished tobacco products if they are sold in final packaging intended for consumer use. FDA intends to enforce the registration and listing requirements under section 905 with respect to such products. Examples of components and parts that are sold or may be sold as finished tobacco products include pipe tobacco filler, filter tubes, e-cigarette batteries, and e-liquids, whether sold separately to consumers or as part of kits.6
Based on our experience with cigarettes, cigarette tobacco, RYO, and smokeless tobacco, we amended our previous compliance policy in July 2016. Under our previous policy, FDA intended to enforce the establishment registration and product listing requirements with
respect to owners and operators engaged in the manufacture of tobacco, paper, filters, and pouches whether or not such products were for further manufacturing of, or for consumer use as, regulated tobacco products. However, FDA announced a change to that policy because we believe we can obtain the necessary information about tobacco product components and parts through other means, such as premarket applications for finished tobacco products and through the use of master files, as explained in the guidance Tobacco Product Master Files.7 Additionally, we aligned our compliance policy for cigarettes, cigarette tobacco, RYO, and smokeless tobacco with the products now regulated as a result of the deeming rule to reduce burden on industry. Should FDA find that additional information is needed to protect the public health, the Agency may reconsider this compliance policy. We intent to communicate any compliance policy changes by guidance or rulemaking.
What Information is Submitted as Part of Establishment Registration and Product Listing Submissions?
Registration Information
Section 905(b) of the FD&C Act sets forth the requirements for submission of establishment registration information. As required by the statute, any person engaged in the manufacturing of tobacco products must register with FDA and submit the following information:
The name and full address of each establishment engaged in manufacturing the registrant owns or operates, as of the date of registration.
The name and places of business of the owner or operator. In the case of a partnership, include the name of each partner. In the case of a corporation, include the name of each corporate officer and director, and the State of incorporation.
The registration and listing electronic submission system and the paper form request additional optional information that FDA also recommends be submitted, including:
An e mail address, to facilitate correspondence between registrants and FDA.
A Data Universal Numbering System (D-U-N-S) number8 or other unique identifier (codes) for the place of business of the owner, the place of business of the operator, and the location of the establishment. The business entity identifier recognized by the FDA Data Standards Council is the D-U-N-S number, and providing the site-specific
D-U-N-S number for an entity will help prevent inaccuracies in FDA’s database.
Product Listing Information
Section 905(i) of the FD&C Act sets forth the requirements for submitting product listing information. The FD&C Act requires that, at the time of registration, the registrant submit a list of all tobacco products that are being manufactured by the registrant for commercial distribution.
The product listing must include certain accompanying information, which will vary depending on the circumstances. These circumstances are as follows:
Under section 905(i)(1)(A), if a tobacco product standard has been established under section 907 of the FD&C Act (21 U.S.C. 387g) with respect to the tobacco product or the tobacco product is subject to premarket review under section 910 of the FD&C Act (21 U.S.C. 387j), then the product listing must include a reference to the authority
for the marketing of the tobacco product and a copy of all labeling for that product. We interpret this to mean that labeling is to be submitted as an exact,
legible, full color copy.
Under section 905(i)(1)(B), the product listing for all other tobacco products must include all labeling for that product. It must also include “a representative sampling of advertisements” for the product. If requested by FDA for good cause, a copy of all advertisements for a particular tobacco product must be submitted. We interpret “a representative sampling of advertisements” to mean typical advertising material that reflects the full range of promotional statements made for the tobacco product. For example, if more than one magazine advertisement is used, but the promotional content is essentially identical, only one need be submitted. In addition, the product listing must include “a copy of all consumer information” to the extent the information is not advertising and has not already been provided as a form of product labeling. Consumer information does not include information directed at wholesalers, distributors, or retailers, where such information is not available to consumers. For example, consumer information does not include product specifications intended for manufacturing purposes, photos of components or parts not intended for individua l sale, or communications between companies.
If a registrant has determined that a product in its product listing is not subject to a tobacco product standard established under section 907 of the FD&C Act, FDA ma y request that the registrant provide a brief statement of the basis for that determination.
We are interpreting section 905(i) of the FD&C Act to require that each product included in a product listing be clearly identified and distinguished. Products that differ in any way, including a difference in any component or part, should be listed separately for purposes of section 905(i). For example, a soft pack and a hard pack of cigarettes should be listed separately, even if the cigarettes included in the pack are identical.
Each product is to be clearly and uniquely identified by the product category9 (e.g., cigarette, smokeless tobacco, paper, filter) and unique name (i.e., brand/sub-brand or other commercial name used in commercial distribution). Tobacco products are to be identified in this way because such names are needed to determine whether products in commercial distribution are
listed as required. You are to include product identification numbers (e.g., SKU, catalog number, UPC) as needed to uniquely identify the product.
However, FDA recognizes that product listing for some tobacco products may result in numerous labeling submissions that the manufacturer must prepare and submit. For example, variations in package size, nicotine strength, Propylene Glycol (PG)/ Vegetable Glycerin (VG) ratio, and flavor can result in thousands of individual product labeling submissions.
In order to reduce the amount of uploaded labeling submissions, FDA does not, at this time, intend to enforce the requirements that owners and operators submit the labeling for each individua l listed tobacco product if the registrant submits information that represents the labeling for a selected line of products. In deciding whether a registrant’s submitted information falls within this compliance policy, FDA may consider whether the tobacco products’ labeling is essentially identical (e.g., the same formatting, fonts, colors, background text, and images) and whether the variations are limited to package size, nicotine strength, PG/VG ratio, and flavor.
However, we recommend that zero nicotine formulas of a product, or product line, be grouped separately from products with nicotine. Appendix A contains an example of how a registrant could submit information that represents the labeling for a selected line of products, and FDA would not intend to enforce the requirements in subsections 905(i)(1)(A) and (B) with regard to labeling submissions.
For any questions regarding this compliance policy, please contact CTPRegistrationandListing@fda.hhs.gov for assistance and include ‘registration and listing labeling submissions’ in the subject line.
Section 905(i)(3) of the FD&C Act requires the following changes to the product list to be reported twice a year:
For any tobacco products you have introduced for commercial distribution and have not included in a previous product listing, the complete product listing information as described above.
For any tobacco products you have discontinued manufacturing for commercial distribution since the last report, notice of the discontinuance containing the established name of the product as previously listed and the date of discontinuance.
For any tobacco products you had given notice as being discontinued and have since resumed manufacturing for commercial distribution, notice of the resumption containing the date of resumption and complete product listing information as described above.
Any material change in any information previously submitted
How to Make Establishment Registration and Product Listing Submissions
The registration and listing electronic submission system is designed to streamline the data entry process for registration and product listing at the Center for Tobacco Products (CTP). Although electronic submission is not required, FDA is strongly encouraging electronic submission to
facilitate efficiency and timeliness of data submission and management. The electronic submission system and FDA Form 3741, an alternative tool for paper submissions, are available at https://www.fda.gov/TobaccoProducts/GuidanceComplianceRegulatoryInformation/Manufacturing/defaul t.htm.
When Must You Register Your Establishment and List Your Products Under Section 905 of the FD&C Act?10
Section 905(b) of the FD&C Act requires establishment registration information to be submitted by December 31, 2009, and to be resubmitted annually on or before December 31st of each year.
Section 905(c) of the FD&C Act requires every person upon first engaging in the manufacturing of a tobacco product in any domestic establishment owned or operated by that person to immediately register. In addition, section 905(d) requires registered owners and operators to immediately register any new establishment that begins manufacturing tobacco products.
Section 905(i)(1) of the FD&C Act requires the complete product list information to be submitted at the time of first registration. In addition, section 905(i)(3) of the Act requires that certain
changes in the product list be submitted biannually, once during June and once during December.
We encourage manufacturers to begin the process as early as possible. Manufacturers may contact FDA to discuss issues relating to registration and listing of their products, such as large volumes of products and labeling files, by emailing FDA at CTPRegistrationandListing@fda.hhs.gov.
This guidance contains information collection provisions that are subject to review by the Office of Management and Budget (OMB) under the Paperwork Reduction Act of 1995 (44 U.S.C. 3501-3520).
The time required to complete this information collection is estimated to average 3.75 hours per response, including the time to review instructions, search existing data sources, gather the data needed, and complete and review the information collection. Send comments regarding this burden estimate or suggestions for reducing this burden to:
Food and Drug Administration Center for Tobacco Products Document Control Center Building 71, Room G335
10903 New Hampshire Avenue Silver Spring, MD 20993-0002
An Agency ma y not conduct or sponsor, and a person is not required to respond to, a collection of information unless it displays a currently valid OMB control number. The OMB control number for this information collection is 0910-0650. To find the current expiration date, search for this OMB control number at https://www.reginfo.gov.
DOCUMENT HISTORY
November 2009 — First edition of guidance issued.
April 2014 — Section III of guidance revised to reflect the change in the electronic submission tool for registration and listing from eSubmitter to a new system available on FDA’s Web page at
https://www.fda.gov/tobacco-products/compliance-enforcement-training/manufacturing and to remove obsolete time frame information.
Throughout section III — The phrase “registration and listing electronic submission system” replaces the phrases “eSubmitter application,” and “FDA eSubmitter tool.”
Section III, page 2 — First paragraph in section III describing eSubmitter tool deleted.
Section III.D, page 7 — Sentences in first paragraph describing eSubmitter tool deleted.
Section III.D, page 7 –Web site address where the electronic submission system and FDA Form 3741 are available was updated.
Section III.D, page 7 — Third paragraph describing how to access the eSubmitter tool deleted.
Section III.E, page 8 — Paragraph describing when to submit information for registration and listing information in 2010 deleted.
Section III.E, page 8 — Paragraph providing November 2009 as the availability date for the eSubmitter tool deleted.
Section IV, page 8 — Expiration date for the information collection updated from 12/31/2012 to 10/31/2015.
May 2014 — Cover page, and section IV, Page 8 — Physical mailing address changed to reflect the center’s relocation to the White Oak Campus.
July 2016 — Registration and listing guidance revised to reflect changes in FDA authorities over “deemed” tobacco products.
Throughout the document — A number of non-substantive changes were made for clarification purposes.
Section II, page 2 — Background updated to reflect changes in FDA authorities over “deemed” tobacco products arising from “deeming” rule.
Section III, page 3 — Accessory, components or part, and finished tobacco product definitions added.
Section IV.B, page 5 — Former Section B “Who Registers and Submits Product Listing Information under Section 905 of the Act” becomes Section A. Compliance policy for cigarettes, cigarette tobacco, RYO, and smokeless tobacco deleted.
Section IV.B, page 6 — “FDA’s Compliance Policy for Regulated Tobacco Products” added; compliance policy for cigarettes, cigarette tobacco, RYO, and smokeless tobacco amended.
Section IV.C, page 6 — Title changed.
Section IV.C, page 7 — Information on Data Universal Numbering System is updated.
Section IV.D, page 9 — Title changed.
Section IV.E page 9 — Title changed.
Section IV.E, page 9 — Last paragraph added to explain when owners and operators of domestic manufacturing establishments engaged in the manufacture of newly deemed tobacco products will need to register and submit product listings.
Section IV.E — Data Universal Numbering System information incorporated to section C.
November 2016 — Section IV.E. revised to: (1) clarify that persons who owned or operated domestic manufacturing establishments that were only engaged in the manufacture of newly deemed products prior to August 8, 2016, but not on or after August 8, 2016, do not have to register with FDA or submit product listing, and (2) explain that FDA does not intend to enforce the requirement to register and submit product listing information by December 31, 2016 for persons who owned or operated domestic manufacturing establishments engaged in the manufacture of newly deemed products prior to August 8, 2016, and continued to own or operate such establishments on or after August 8, 2016, provided the submissions are received by FDA on or before June 30, 2017.
September 2017 — Section II added clarifications to background section. Sections IV.C.2 revised to provide compliance policy for certain product listing labeling submissions.
Section IV.E amended compliance date to September 30, 2017.
September 2017 — Section IV.E amended compliance date to October 12, 2017.
December 2017 — Section IV.E amended to provide compliance policy for product listing updates for December 2017 with respect to the manufacture of newly deemed tobacco products provided they registered and listed those products by October 12, 2017.
MONTH 2023—Registration and listing guidance revised to reflect changes in the statutory definition of “tobacco product” and FDA authority over products containing nicotine not made or derived from tobacco.
Section III - definition of “tobacco product” is updated to reflect statutory amendments made by the Consolidated Appropriations Act, 2022 (Pub. L. 117-103). Among other things, the legislation amends the definition of “tobacco product” in section 201(rr) of the FD&C Act to include products “containing nicotine from any source.”
Section IV.E— outdated compliance policy for deemed tobacco products removed. Footnote 10 revised to note applicability of section 905 of the FD&C Act to tobacco products containing non-tobacco nicotine.
APPENDIX A –EXAMPLE PACKAGE LABEL PLAN

Note: Appendix A is intended to serve as one example of a circumstance in which FDA does not intend to enforce the requirements in subsections 905(i)(1)(A) and (B) with regard to labeling submissions. It describes a “package label plan,” which could be submitted by a registrant to represent the labeling for a selected line of products. FDA may exercise enforcement discretion in other circumstances as described in the guidance.
In this example, a package label plan consists of two parts submitted together, a “model label” and a “product variation index.”
The model label contains a proxy, such as placeholder text, for the variations in the label, such as variations in package size, nicotine strength, PG/VG ratio, and flavor. The proxy, such as placeholder text, is the same size, font, and color as it will appear on the actual label for the individual product. All formatting, fonts, colors, background text, and images are represented on the model label as they will appear on the actual label.
The product variation index is a listing of all variations for a specific tobacco product; limited to variations in package size, nicotine strength, PG/VG ratio, and flavor. A product variation index lists all combinations of the variations that will be using the model label. Each tobacco product listing on the index corresponds with the attached model label. The product variation index indicates the corresponding model label and also includes product name and product identification number columns.
The package label plan would be uploaded to the electronic submission portal, as discussed in Section IV.D of the guidance, and be associated with all product listings represented in the variation index. For the products using a particular package label plan, the registrant would upload one file and that file would be associated with the relevant product listings by selecting those listings. Associating a label submission with a product that does not use that label or is not covered by that package label plan would not fall within the above stated compliance policy.
Upon reviewing the package label plan, if FDA determined that it needed additional labeling submissions, it would notify the firm that the submission of additional labeling is needed. In addition, the package labeling must comply with all other applicable labeling requirements under the FD&C Act and implementing regulations.
The examples below are for ‘Generic E-Juice’ an e-liquid product, which comes in three package sizes, three different nicotine strengths, including a zero nicotine formula, two variations of PG/VG ratio, and three flavors.
Example A:
I.
Sample
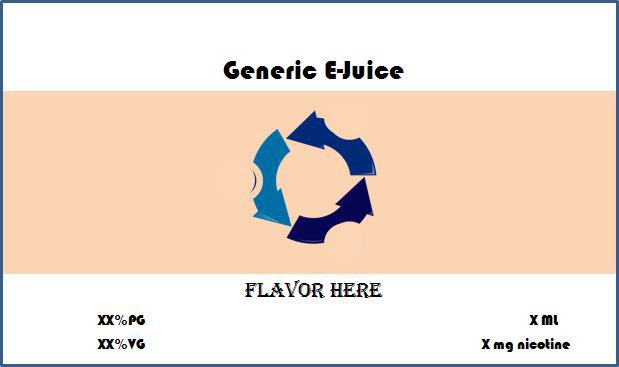
Note: In the above model label, the proxy for package size, nicotine strength, PG/VG ratio and flavor are all being represented by placeholder text. Here, the variable elements are represented by X’s and the text ‘Flavor Here’.
Formatting, fonts, colors, background text, and images, and any other elements except package size, nicotine strength, PG/VG ratio, and flavor should remain identical across labels for all products listed under this package label plan. All proxies, including placeholder text, should be represented in the font, size, and color in which they will appear on the actual label.
II. Product Variation Index
Product Name |
Product Identification Number |
Size |
Nicotine Strength |
PG/VG Ratio |
Flavor |
Generic E-Juice |
G001 |
5ml |
3mg |
50%PG/50%VG |
A |
Generic E-Juice |
G002 |
5ml |
3mg |
50%PG/50%VG |
B |
Generic E-Juice |
G003 |
5ml |
3mg |
50%PG/50%VG |
C |
Generic E-Juice |
G004 |
5ml |
3mg |
70%PG/30%VG |
A |
Generic E-Juice |
G005 |
5ml |
3mg |
70%PG/30%VG |
B |
Generic E-Juice |
G006 |
5ml |
3mg |
70%PG/30%VG |
C |
Generic E-Juice |
G007 |
5ml |
6mg |
50%PG/50%VG |
A |
Generic E-Juice |
G008 |
5ml |
6mg |
50%PG/50%VG |
B |
Generic E-Juice |
G009 |
5ml |
6mg |
50%PG/50%VG |
C |
Generic E-Juice |
G010 |
5ml |
6mg |
70%PG/30%VG |
A |
Generic E-Juice |
G011 |
5ml |
6mg |
70%PG/30%VG |
B |
Generic E-Juice |
G012 |
5ml |
6mg |
70%PG/30%VG |
C |
Generic E-Juice |
G013 |
10ml |
3mg |
50%PG/50%VG |
A |
Generic E-Juice |
G014 |
10ml |
3mg |
50%PG/50%VG |
B |
Generic E-Juice |
G015 |
10ml |
3mg |
50%PG/50%VG |
C |
Generic E-Juice |
G016 |
10ml |
3mg |
70%PG/30%VG |
A |
Generic E-Juice |
G017 |
10ml |
3mg |
70%PG/30%VG |
B |
Generic E-Juice |
G018 |
10ml |
3mg |
70%PG/30%VG |
C |
Generic E-Juice |
G019 |
10ml |
6mg |
50%PG/50%VG |
A |
Generic E-Juice |
G020 |
10ml |
6mg |
50%PG/50%VG |
B |
Generic E-Juice |
G021 |
10ml |
6mg |
50%PG/50%VG |
C |
Generic E-Juice |
G022 |
10ml |
6mg |
70%PG/30%VG |
A |
Generic E-Juice |
G023 |
10ml |
6mg |
70%PG/30%VG |
B |
Generic E-Juice |
G024 |
10ml |
6mg |
70%PG/30%VG |
C |
Generic E-Juice |
G025 |
15ml |
3mg |
50%PG/50%VG |
A |
Generic E-Juice |
G026 |
15ml |
3mg |
50%PG/50%VG |
B |
Generic E-Juice |
G027 |
15ml |
3mg |
50%PG/50%VG |
C |
Generic E-Juice |
G028 |
15ml |
3mg |
70%PG/30%VG |
A |
Generic E-Juice |
G029 |
15ml |
3mg |
70%PG/30%VG |
B |
Generic E-Juice |
G030 |
15ml |
3mg |
70%PG/30%VG |
C |
Generic E-Juice |
G031 |
15ml |
6mg |
50%PG/50%VG |
A |
Generic E-Juice |
G032 |
15ml |
6mg |
50%PG/50%VG |
B |
Generic E-Juice |
G033 |
15ml |
6mg |
50%PG/50%VG |
C |
Generic E-Juice |
G034 |
15ml |
6mg |
70%PG/30%VG |
A |
Generic E-Juice |
G035 |
15ml |
6mg |
70%PG/30%VG |
B |
Generic E-Juice |
G036 |
15ml |
6mg |
70%PG/30%VG |
C |
Example B:
I.
Sample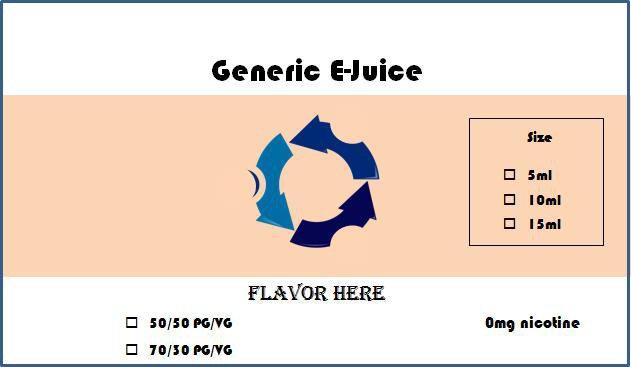
Note: In the above model label for the zero nicotine formulation, the proxy for size and PG/VG ratio are represented with unmarked checkboxes and the flavor is indicated with the placeholder text “Flavor Here.”
Formatting, fonts, colors, background text, and images, and any other elements except package size, PG/VG ratio, and flavor, should remain identical across labels for all products listed under this package label plan. All proxies, including placeholder text, should be represented in the font, size, and color in which they will appear on the actual label.
II. Product Variation Index
Product Name |
Product Identification Number |
Size |
Nicotine Strength |
PG/VG Ratio |
Flavor |
Generic E-Juice |
G037 |
5ml |
0mg |
50%PG/50%VG |
A |
Generic E-Juice |
G038 |
5ml |
0mg |
50%PG/50%VG |
B |
Generic E-Juice |
G039 |
5ml |
0mg |
50%PG/50%VG |
C |
Generic E-Juice |
G040 |
5ml |
0mg |
70%PG/30%VG |
A |
Generic E-Juice |
G041 |
5ml |
0mg |
70%PG/30%VG |
B |
Generic E-Juice |
G042 |
5ml |
0mg |
70%PG/30%VG |
C |
Generic E-Juice |
G043 |
10ml |
0mg |
50%PG/50%VG |
A |
Generic E-Juice |
G044 |
10ml |
0mg |
50%PG/50%VG |
B |
Generic E-Juice |
G045 |
10ml |
0mg |
50%PG/50%VG |
C |
Generic E-Juice |
G046 |
10ml |
0mg |
70%PG/30%VG |
A |
Generic E-Juice |
G047 |
10ml |
0mg |
70%PG/30%VG |
B |
Generic E-Juice |
G048 |
10ml |
0mg |
70%PG/30%VG |
C |
Generic E-Juice |
G049 |
15ml |
0mg |
50%PG/50%VG |
A |
Generic E-Juice |
G050 |
15ml |
0mg |
50%PG/50%VG |
B |
Generic E-Juice |
G051 |
15ml |
0mg |
50%PG/50%VG |
C |
Generic E-Juice |
G052 |
15ml |
0mg |
70%PG/30%VG |
A |
Generic E-Juice |
G053 |
15ml |
0mg |
70%PG/30%VG |
B |
Generic E-Juice |
G054 |
15ml |
0mg |
70%PG/30%VG |
C |
1 This guidance was prepared by the Office of Regulations and the Office of Compliance and Enforcement in the Center for Tobacco Products at FDA.
2 Foreign establishments are not required to register and list until FDA issues regulations establishing such requirements in accordance with section 905(h) of the FD&C Act (21U.S.C. 387e(h)).
3 Accessories of tobacco products subject to the deeming rule are explicitly excluded from the rule’s deeming provision.
4 Examples of currently marketed products that are subject to the deeming rule include: cigars, pipe tobacco, nicotine gel, dissolvable nicotine products, and electronic nicotine delivery systems (“ENDS”), including electronic cigarettes (also known as e-cigarettes or e-cigs), e-hookah, e-cigars, vape pens, personal vaporizers (also known as advanced personal vaporizers or APVs),electronic pipes, and nicotine-containing liquids, including the e-liquids used with ENDS products, among other products. For further information on compliance deadlines for requirements under the deeming rule, see FDA guidance for industry Extension of Certain Tobacco Product Compliance Deadlines Related to the Final Deeming Rule, available at https://www.fda.gov/downloads/TobaccoProducts/Labeling/RulesRegulationsGuidance/UCM557716.pdf.
5 However, and as explained above, accessories of tobacco products subject to the deeming rule are explicitly excluded from the rule’s deeming provision. Thus, although they meet the definition of tobacco product, such accessories are not currently subject to regulation under the FD&C Act (including section 905). See below for details on FDA’s compliance policy for tobacco products that are sold or distributed solely for further manufacturing.
6 As explained above accessories of deemed tobacco products are not currently subject to section 905.
7This guidance is available on the CTP guidance Web page at http://www.fda.gov/TobaccoProducts/Labeling/RulesRegulationsGuidance/default.htm.
8 D-U-N-S numbers are proprietary to and controlled by Dun & Bradstreet. If the D-U-N-S number for a location has not been assigned, a business may obtain one for no cost directly from Dun & Bradstreet (http://www.dnb.com). Please note that registrants who wish to obtain a new D-U-N-S number should obtain one well in advance of FDA’s deadline, because it may take 30 days (or longer) to process a new number. Please note that the D-U-N-S Number is not required. Alternatively, you may elect to receive a D-U-N-S number within one business day by paying a fee. Please consult Dun & Bradstreet (http://www.dnb.com) directly for more information.
9 At this time, some product categories used for product listing may fall under a subcategory for a premarket tobacco product submission (e.g., a filter is a category for listing purposes, but a subcategory under roll your own for a premarket submission).
10 On April 14, 2022, owners and operators of establishments engaged in the manufacture, preparation, compounding, or processing of tobacco products containing nicotine not made or derived from tobacco became subject to the requirements of section 905 of the FD&C Act described in this section.
| File Type | application/vnd.openxmlformats-officedocument.wordprocessingml.document |
| File Title | Registration and Product Listing for Owners and Operators of Domestic Tobacco Product Establishments |
| Subject | Guidance for Industry |
| Author | FDA/Center for Tobacco Products |
| File Modified | 0000-00-00 |
| File Created | 2023-08-24 |
© 2026 OMB.report | Privacy Policy