Protocol
Data To Support Social and Behavioral Research as Used by the Food and Drug Administration
Protocol
OMB: 0910-0847
OMB Control No.: 0910-0847
Expiration Date: 12/31/2022

Training Decay Selection for Medical Products Pilot Human Factors Study
PROTOCOL
BBA Number: EETWP26
Document Number: UserWiseFDA01
Version 03
Revision History
Version # |
Date |
Reasoning and Description of Changes |
Name |
01 |
05AUG2019 |
Initial document version issued to FDA for review. |
L. Sneath |
02 |
11SEP2019 |
Document updated based on FDA feedback. |
L. Sneath |
03 |
14NOV2019 |
Document updated based on FDA feedback. |
A. Kristedja |
Table of Contents
1 References 5
2 Abbreviations and Definitions 6
2.1 Abbreviations 6
2.2 Definitions 6
3 Introduction 8
3.1 Study Background 8
3.2 Objectives 9
3.3 Study Design Overview 10
4 Product Description 11
4.1 Insulin Pump 11
4.1.1 Insulin Pump High-Level Task Analysis 13
4.1.2 Study Articles and Equipment 14
4.2 Intended Use Environment 15
4.3 Simulated Use Environments 15
5 Study Personnel 16
5.1 Training of Study Personnel 16
6 Users 17
6.1 Intended Users 17
6.2 Recruitment 17
6.3 Participant Cohort Size Calculations 18
6.3.1 Literature Review and Effect Size Determination 18
6.3.2 Pilot Study Participant Cohort Size 20
6.3.3 Main Study Participant Cohort Size 21
6.4 Participant Cohort Randomization 21
6.5 Payments to Participants 22
7 Study Design 22
7.1 Study Session Design 22
7.1.1 Introduction and Consent 23
7.1.6 End of Session Questionnaire 27
7.2 Task Type Categorization 28
7.3 Risk Assessment Summary 29
7.4 Methods for Capturing Use Errors 38
7.5 Data Recording and Assessment 39
7.6 Task Assessments and Rating Criteria 40
7.7 Final Analysis and Acceptance Criteria 41
7.7.1 Statistical Analyses Plan 41
8 Additional Study Information 42
8.1 Informed Consent 42
8.2 Ethical Considerations 42
8.3 Requirements of HIPAA 43
8.4 Safety Reporting to Regulatory Authorities, Investigators, and IRBs 44
List of Tables
Table 2: List of Abbreviations 6
Table 3: List of Definitions 6
List of Figures
Figure 1: Medtronic Minimed Insulin Pump Overview 12
Figure 2: Quickset Infusion Set 12
Figure 4: Minimed Quick Serter (Optional) 13
Figure 5: Example of an Insulin Vial 13
Document No. |
Document Name |
- |
Arthur, W., Bennett, W., Edens, P. S., & Bell, S. T. (2003). Effectiveness of training in organizations: A meta-analysis of design and evaluation features. Journal of Applied Psychology, 88(2), 234–245. |
- |
Champely, S. (2018). pwr: Basic Functions for Power Analysis. Retrieved from https://CRAN.R-project.org/package=pwr |
- |
Clark, S. (2016). Training Decay Selection for Human factors Validation. |
- |
FDA Guidance (2016). Applying Human Factors and Human factors Engineering to Medical Devices. |
- |
FDA (Draft, 2016). List of Highest Priority Devices for Human Factors Review. |
- |
FDA Guidance (2014). Infusion Pumps Total Product Life Cycle. |
- |
Kester L., Kirschner P.A. (2012) Cognitive Tasks and Learning. In: Seel N.M. (eds) Encyclopedia of the Sciences of Learning. Springer, Boston, MA |
- |
Mead, S. E., & Fisk, A. D. (1997). Effects of Matching Cognitive and Perceptual-Motor Training to Task Components on Complex Task Performance by Older and Younger Adults. Proceedings of the Human Factors and Ergonomics Society Annual Meeting, 41(1), 115–119. |
- |
Meador, Douglas, and Hill, Raymond (2008). Modeling Training Effects on Task Performance Using a Human Performance Taxonomy. ProQuest Dissertations and Theses. |
- |
Zhang, Yi, et al. (2010). A Hazard Analysis for a Generic Insulin Infusion Pump. Journal of Diabetes Science and Technology, Diabetes Technology Society. |
- |
Weiss, B.D., et al. (2005). Quick assessment of literacy in primary care: the newest vital sign. Annals of Family Medicine, 3(6), 514-522. |
ISO 14971:2012 |
Medical devices -- Application of risk management to medical devices |
IEC 62366-1:2015 |
Medical devices - Application of human factors engineering to medical devices |
EN ISO 14971:2012 |
Medical devices – Application of risk management to medical devices |
6025306-019_a |
Medtronic Minimed Paradigm REAL-TIME Revel Insulin Pump User Guide |
UserWiseFDA02 |
Training Decay Selection Human Factors Study – Initial Training Script (Insulin Pump) |
UserWiseFDA03 |
Training Decay Selection Human Factors Study – Moderator’s Script (Insulin Pump) |
UserWiseFDA04 |
Training Decay Selection Human Factors Study – Participant Screener |
UserWiseFDA05 |
Training Decay Selection Human Factors Study – End of Study Questionnaire |
UserWiseFDA06 |
Training Decay Selection Human Factors Study – Observer Datasheet |
UserWiseFDA07 |
Insulin Pump Use-Related Risk Analysis |
UserWiseFDA08 |
Training Decay Selection Human Factors Study – Informed Consent Form for Untrained Participants |
UserWiseFDA09 |
Training Decay Selection Human Factors Study – Informed Consent Form for Trained Participants |
Table 2: List of Abbreviations
Abbreviation |
Definition |
AAMI |
Association for the Advancement of Medical Instrumentation |
ANSI |
American National Standards Institute |
FDA |
Food and Drug Administration |
uFMEA |
Use-Related Failure Modes and Effects Analysis |
ICF |
Informed Consent Form |
ICH |
The International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use |
IRB |
Institutional Review Board |
ISO |
International Standards Organization |
PCA |
Perception, Cognition, Action |
Portable Document File |
|
UM |
User Manual |
Term |
Definition |
Acceptance Criteria |
Criteria that, when met, demonstrate that risks related to human factors have been controlled to acceptable levels. |
Close Call |
The user has difficulty or makes a use error, but the user takes an action to recover and prevents the harm from occurring (i.e., near-miss). |
Critical Task |
A user task which, if performed incorrectly or not performed at all, would or could cause harm to the patient or user, where harm is defined to include compromised medical care. |
Critical Use Error |
User action or lack of action that was different from that expected by the manufacturer and caused a result that (1) was different from the result expected by the user and (2) was not caused solely by device failure and (3) did or could result in harm. |
Difficulty |
The user is able to complete the task safely and effectively but had significant hesitation or challenges while completing the task. |
Objective Data |
Data collected through direct observation. |
Study Article |
Medical device (and medical device accessories) under evaluation. |
Study Equipment |
Third-party medical devices and supplies required to use the Study Article. |
Subjective Data |
Data collected through user feedback via comments, ratings, and assessments provided by participants. |
Training Decay |
A function of loss of skill or retention of information over time. For the purpose of this study, the training decay length is defined as the length of time between the end of the baseline human factors evaluation performed after training until the start of the next human factors evaluation session. |
Use Error1 |
User action or lack of user action, while using the medical device, that leads to a different result than that intended by the manufacturer or expected by the user. |
User Interface |
All means of interaction between the medical device and the user, including both hardware and software interfaces. |
This protocol defines the methodology for a pilot human factors (HF) study and main human factors study of a medical device to evaluate the effect of various training decay lengths and skill types on user performance. An infusion pump was selected as the medical device to evaluate for this research study for its complexity and inclusion of tasks that require various skill types.
This research study is sponsored by the Food and Drug Administration (FDA) and will be performed in the U.S. The study will be conducted by UserWise, Inc., an independent consultancy, based in the U.S., which specializes in the human factors and human factors of medical technologies, as well as San Jose State University (SJSU). The Moderator(s) and Observer(s) will be UserWise, Inc. personnel, who are experienced Human Factors Engineers, or Principal Investigator (PI) approved SJSU personnel.
The pilot human factors study may uncover changes that need to be made to the study methodology or approach. If changes to the protocol are required, based on the findings from the pilot human factors study, the revised document will be reviewed/approved by the appropriate FDA and IRB stakeholders prior to the start of the main human factors study.
The Food and Drug Administration (FDA) requires high-fidelity simulated use (i.e., human factors (HF) validation) of a medical device by intended users, prior to marketing medical devices and combination products in the United States. When conducting HF validation testing, representative users must use the device under the expected use conditions in the field. Some medical products require training prior to use. In order to realistically simulate use in these cases, it is necessary to provide representative device training as part of the device HF validation study.
When the FDA requires training to be “representative” in an HF validation study, it means that the training offered during testing must be equivalent to the training that will be offered in the real world. As part of ensuring “representativeness,” the participant should not be tested on the use of the device immediately after being trained unless training would normally occur directly before the initial use of the device. In the real world, there is usually a period of days or even weeks between the point in time a user is trained and the moment they use the device for the first time. For this reason, the FDA acknowledges the need for considering “training decay” as part of HF validation testing.
While the amount of time required to ensure adequate training decay is debated among human factors practitioners, simulating training decay as part of the human factors study is practiced throughout the medical device industry. Simulating training decay requires some manufacturers to train the study participants, send them home (if the training decay period is long enough), and then invite them back after a period of time for HF validation testing. In some cases, the need for simulating a longer training decay can be associated with drawbacks in the following scenarios:
Use of specialty surgeons from around the country: training decay may reduce the sponsor’s ability to recruit from diverse institutions. For example, testing of a medical device might need to be limited to a single hospital site because of the logistics concern of ensuring the return of participants in order to fully test the effect of training decay and, thus, precluding a more representative multi-site plan.
Products with unique user populations: simulating training decay may reduce the sponsor’s ability to recruit enough users who can return for retesting.
Evaluations involving a team of participants together as a group (e.g., An Operating Room Team): with team evaluations, it is important that all participants return as a group. If the teams returning are mixed, this may introduce a significant number of study artifacts and reduce the validity of the study with respect to simulating a team of co-workers in a hospital.
Overall reduced number of participants who will actually return for the HF validation study: due to the increased time commitment with training decay simulation, the study population may be unrepresentative, due to self-selection bias.
In response to these challenges, medical device manufacturers typically simulate shortened training decay periods. UserWise, Inc. conducted an informal survey of four (4) manufacturers that develop injection devices and found that it is typical to have 2-week real-time training decay simulated in HF validation studies by using abbreviated lengths of testing delay time vary greatly, ranging between only 1 day to a full 2 weeks.
Variability in how manufacturers simulate training decay is due, in part, to the lack of concrete methodology for identifying an appropriate and suitable training decay period.
The primary goal of this training decay research study is to identify preliminary results to identify generalizable training decay curves. The objectives of the training decay research that will be explored in this human factors study are to:
Quantify training decay curves, and
Examine differences in decay between task types.
The research outlined in this protocol aims to identify reliable and replicable training decay behavior to identify how generalizable training decay curves could standardize the methods for determining the appropriate length of training decay, while conducting human factors testing for medical products. Standardizing this may ultimately improve use error identification, while avoiding an undue toll on manufacturer resources and delays in getting life improving innovative products to patients.
This protocol defines the methodology for conducting the training decay research with an objective of revealing information on training decay curves associated with training on the use of a medical product. The research aims to identify generalizable training decay curves.
As secondary research endpoints, the influence of task difficulty and task type (e.g., cognitive, psychomotor/motor, and perceptual) on training decay curves will be evaluated. Participants will be asked to rank the difficulty of tasks on a Likert scale at the end of their last test session (see Section 7.1.6.2). This information will be analyzed, using the statistical methods described in Section 7.7.1.
Additionally, as time allows, information will be gathered to inform the effect of “accelerated decay” by asking participants in the trained cohorts:
how stressed they felt at the start of the baseline assessment and the main evaluation session
how tired they felt at the start of the baseline assessment and the main evaluation session
how they spent their time during the training decay period (for the one-hour training decay cohort, only)
After the study, the relationship between how participants spent their time and their performance will be reviewed at a high level to identify whether various activities or stress levels may have “accelerated” the decay. This is a tertiary research endpoint, and this information will be assessed only at a high-level as this study is not designed to thoroughly evaluated the effects of “accelerated decay.”
In a representative use environment, trained participants will be provided with training that is representative of training currently used in human factors validation testing of medical products for FDA clearance/approval. The training will include a training video and/or a hands-on component, following the steps in the User Manual. Participants will have the opportunity to ask a trained moderator clarifying questions at the end of the training. The overall length of this training session as well any clarifying questions will be recorded. Once training is completed, the participants will be assessed on their ability to perform the tasks on which they were just trained with no training decay. Task success will be collected for each task as outlined in the Moderator’s script/datasheet, which will allow the Moderator(s) and Observer(s) to quantify which steps a participant completes correctly. This assessment will be used as a within-participant baseline assessment with which future timepoints can be compared.
To address the goal of quantifying training decay curves, participants will be assigned to one of four cohorts, specifying various lengths of training decay. Depending on their cohort assignment, participants will be asked to come back in one hour, one day, or one week to complete the follow-up assessment, representing one-hour, one-day, or one-week training decays, respectively. Some imprecision, regarding exactly when they return for the second (main) assessment. will be allowed for the sake of practicality. One cohort will be assigned no training to simulate “infinite training decay” and, thus, will have no associated training decay period.
After training and the designated training decay period (based on cohort assignment indicated in Section 6.3), participants in the one-day or one-week training decay cohort will return to the study site for a second human factors study session. Participants in the one-hour training decay cohort will not need to return, as they will be asked to remain near the study room, during their break. Participants in the no training cohort will not need to return to the study site, as they will only participate in one study session. Upon the start of the second session (as applicable), participants will receive a brief overview of the purpose of the study, provided by the Study Moderator, and then will be provided with the product and all items needed to successfully use the product and will be asked to use the product. The same script will be used for both study sessions; therefore, participants will perform the same task with the device that they had done before the training decay period. User performance will be recorded, using the same methods employed in the baseline assessment. Task success will be collected for each of the Critical and Essential Tasks as outlined in Section 7.3 to quantify which steps a participant completes correctly. Results will be compared to their previously recorded baseline performance to quantify the effects of various training decay periods.
A pilot study with twelve (12) participants will first be conducted to inform the study design. After the pilot study results have been analyzed, the data obtained will be used to inform whether any changes are required to the study design and/or documents, prior to execution of the main human factors study (referred to as the “main” human factors study). This protocol is the test plan for both the pilot study and the main human factors study; sections specific to only the pilot study or only the main study are noted as such in the section title. If changes to the protocol are required, based on the findings from the pilot study, the revised document will be reviewed/approved by the appropriate FDA and IRB stakeholders, prior to the start of the main human factors study.
To address the goal of examining differences between task types, the Medtronic Minimed Paradigm Real-Time Revel insulin pump, Model Number MMT-523, was selected to be fairly difficult to allow for a range of success/failures and, therefore, allow for differences in training decay between task types to be identified. The device was selected, based on input from industry and FDA stakeholders.
Many tasks related to setting up the reservoir and infusion set involve psychomotor/motor skills; whereas, there are other tasks which require cognitive skills (e.g., knowing the difference between a basal and bolus dose or remembering to wipe the injection site with alcohol) or perceptual skills (e.g., seeing and comprehending a prompt on the infusion pump screen). Categorizing each task into task types will allow for the hypotheses on training decay to be based on literature involving relevant cognitive processes as well as increase the potential to generalize the results to similar devices. See Section 7.2 for further details on the task type categories intended to be used for this research.
The Minimed insulin pump (Figure 1) is a series of insulin pumps manufactured by Medtronic for patients with diabetes mellitus. The pump operates with a single 1.5 V AAA alkaline battery. As a safety measure, the pump is designed to only accept a new battery and will trigger a failed battery test alarm if the battery has been depleted below the acceptable threshold.
The insulin pump is used in conjunction with the Minimed QuickSet Infusion Set (Figure 2) and Minimed Reservoir (Figure 3) to deliver the programmed insulin dose to the patient. The insulin in the vial (Figure 5) is first transferred to the reservoir by connecting the transfer guard to the vial. The adhesive end of the infusion set may be attached by hand or with the assistance of the Quick Serter (Figure 4). Once the filled reservoir is attached to the infusion set and inserted into the insulin infusion pump, the pump uses a piston-plunger pump to infuse a programmed amount of insulin into the patient through the infusion set tubing.
The Paradigm series of insulin pumps has the option to use a one-way wireless radio frequency link to receive blood sugar measurements from select glucose meters; however, this feature will not be included within the scope of this study.

Figure 1: Medtronic Minimed Insulin Pump Overview
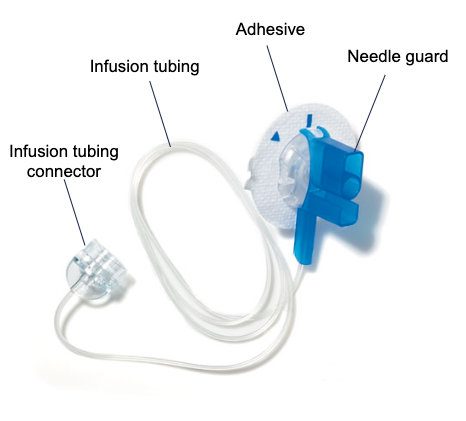
Figure 2: Quickset Infusion Set

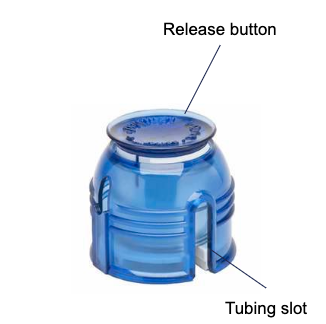
Figure 4: Minimed Quick Serter (Optional)
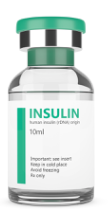
Figure 5: Example of an Insulin Vial
A task analysis (in the form of a workflow, shown in Figure 6) and a use-related risk analysis were developed to identify the tasks associated with using the insulin pump. All tasks shown in Figure 6 will be evaluated in the human factors study.
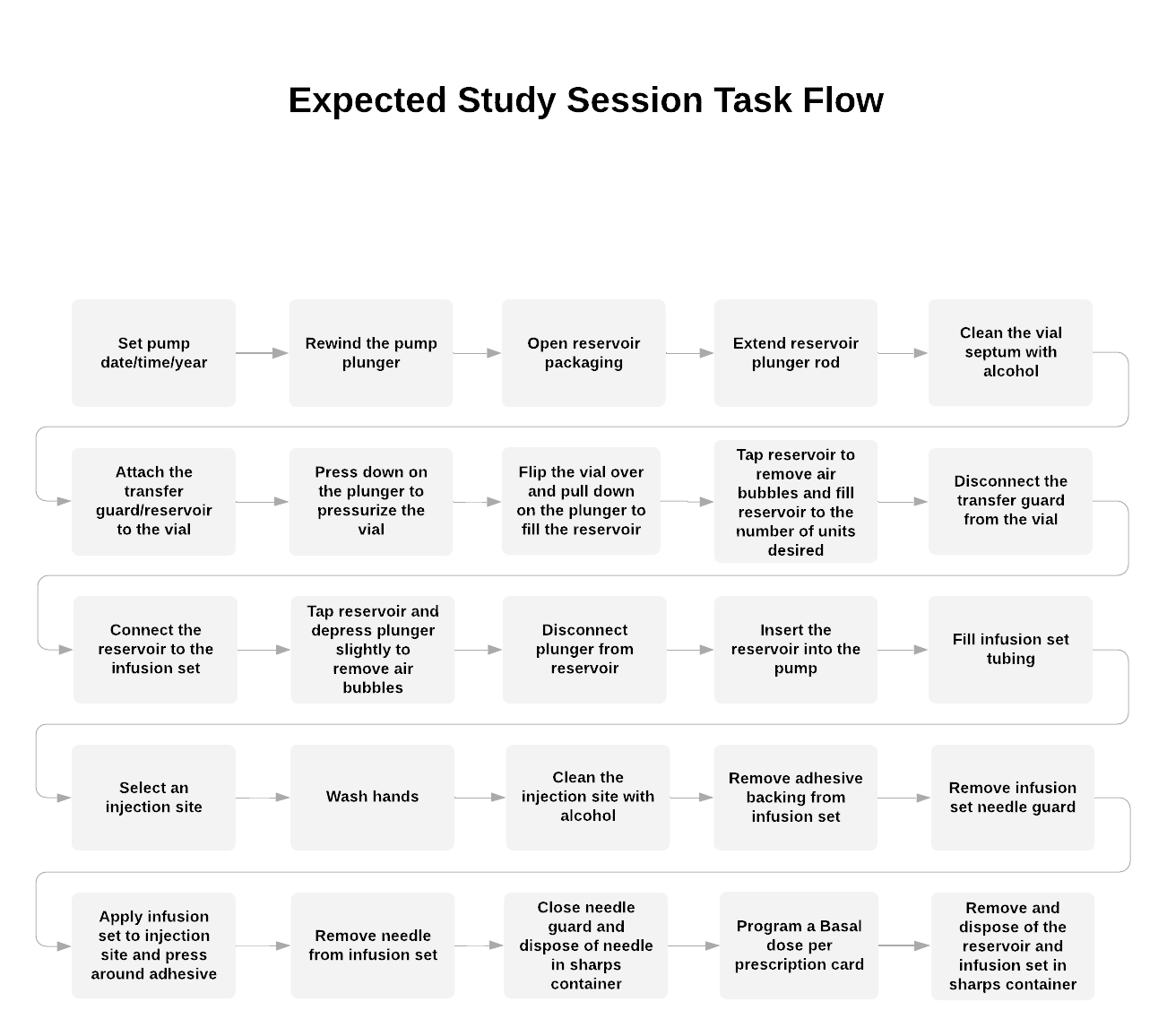
Figure 6: Workflow Tasks for the Minimed Insulin Pump
Study Articles:
Medtronic Minimed Insulin Pump (“Insulin Pump”)
Medtronic Minimed QuickSet Infusion Set (“Infusion Set”)
Medtronic Minimed 1.8 mL Reservoir (“Reservoir”)
Medtronic Minimed Quick Serter Insertion Device (“Insertion Device”)
Vial (filled with sterile water for injection)
Note: The trademark names “Medtronic,” “Minimed,” “Quick Serter,” or others associated with the Medtronic brand will be redacted from the study materials, study articles, and equipment to the degree possible by the study team to decrease potential bias. Branding is not relevant to the research study and, therefore, will be redacted, wherever possible. The verbiage for the various components being used for the participants in the study sessions are noted above in parentheses.
Other standard equipment and materials that will be available during study sessions include:
Alcohol wipes
A trash can
A sharps container
Hand sanitizer
Cotton balls
Clean towels
A simulated sink (with soap and paper towels)
A phone (to call “Customer Service”)
Exam gloves
A complete list of the equipment and accessories used will be documented in the report. In addition, a complete list of the study articles used with part numbers, as available, will be documented in the study report.
The Minimed Paradigm Revel insulin pumps are indicated for the continuous delivery of insulin, at set and variable rates, for the management of diabetes mellitus in persons requiring insulin.
The medical product selected, an insulin pump, will be used primarily in an in-home setting, at the HCP’s office, and in the workplace with normal light and noise conditions.
On occasion, it may be used in a noisy or distracting environment although this is unlikely.
Space constraints, lighting, and noise are examples of environmental factors that affect user performance (U.S. Food and Drug Administration (FDA), 2016). Therefore, the conditions under which simulated use will take place and human factors will be tested shall be replicated.
The study will be performed in an environment that is representative of a home environment. Attention will be paid to relevant factors that may affect user performance:
Representative lighting levels
Representative ambient noise (in this case, a relatively quiet environment that is free from distracting noises)
Space constraints, including limited surface spaces on which to work, shall be simulated via the use of representative furniture
A couch with a coffee table will be included in the study environment to simulate a living room and also to allow adequate working space for the product and User Manual. This configuration was also selected to allow an unobstructed view of the participant’s interactions with the product and instructions.
On this basis, the results of testing may be generalized to anticipated actual use environments.
The study will be conducted on the premises of San Jose State University in the Human Factors Engineering Laboratory, Room 494, in the Engineering Building. The study location and exact dates will be recorded in the study report.
The following roles will be fulfilled by either San Jose State University personnel and/or UserWise, Inc. consultants, during the study:
Study Moderator(s) – responsible for consenting the participant, moderating the study, guiding the participant through the simulated tasks, and asking questions to probe about observed use errors, hazardous situations, and difficulties (as time permits). The Moderator may also take notes, during the study sessions, and answer participant questions at the end of training, as needed.
Study Facilitator(s) – responsible for organizing the simulated environment, study articles, equipment, and required documentation (e.g., non-disclosure agreements).
Study Observer(s) – responsible for taking notes, during the study sessions. The Observer may also answer participant questions at the end of training, as applicable.
Individuals fulfilling the roles above will be recorded in the study report.
The Study Moderator(s) and Observer(s) will train on the medical product, using materials available from the device manufacturer (i.e., online video training, guides, etc.) and the User Manual. The trainings expected to be completed before the start of any study session for various personnel are listed in Table 4.
Training |
Source of Training |
Required for |
Protecting Human Subject Research Participants Training |
Online via Collaborative Institute Training Initiative (CITI Program) |
Primary Investigator(s), Moderator(s), Observer(s), personnel involved in recruitment |
Moderator Training |
Instructor-led by qualified PI-approved personnel |
Moderator(s) |
Project Team Training (including a review of the study scope, methodology, and success/failure criteria for each task) |
Instructor-led by qualified PI-approved personnel |
Moderator(s), Observer(s), Facilitator(s) |
Specific details, regarding the trainings provided and methods used to train the Study Moderator(s), Observer(s), and Facilitator(s), will be included in the study report.
The scope of this research is limited to testing healthy adults without severe mental, physical, or psychological disabilities that would present a significant barrier to using a medical device as the intended user group.
To ensure the participants fit the intended user group of healthy adults, participants will self-report in the recruitment screener if they have a severe mental, physical, or psychological disability that may present a significant barrier to using a medical device. Participants who do not fit this inclusion criteria will be disqualified (see Section 6.2.1 for additional information on inclusion criteria). For additional data, after the study, session participants will be asked to complete the Newest Vital Sign (NVS) test and provide their education level, both of which will inform cognitive ability (see Section 7.1.6 for additional information on the end of session questionnaire).
Lay users will be selected to reflect the general population of the United States.
Participants in the study will be selected to be representative of the general population of the United States and fulfill the required characteristics noted in Section 6.2.1. Participants with a wide variety of educational background and professions will be recruited for this study.
Participants will be recruited, using industry standard and IRB-approved methods
Participants will be told that they are participating in a medical product human factors study. They will be assigned to their cohort, following a randomization scheme, prior to scheduling the baseline visit and follow-up visit. To reduce potential drop-outs, participants will be made aware, before they are scheduled, of whether they are invited to participate in one or two study sessions (depending on the cohort to which they are assigned) and asked to confirm availability for both sessions. Participants will be unaware that there are other participant cohorts with different return times in the study to avoid participant bias.
Using their experience recruiting college and non-college student populations, San Jose State University will make an effort to recruit, such that our sample population mirrors the U.S. adult population.
Only participants who meet the following requirements will be enrolled into the human factors study:
Is 18 years or older
Currently lives in the United States
Can read and comprehend written and spoken English
Does not have a severe phobia of needles, as needles will be present and used during the study sessions. Use of needles and potential risks are disclosed in the Informed Consent Form
Is not pregnant and does not expect to be pregnant soon
Does not suffer from a severe mental, physical, or psychological disability that would present a significant barrier to using a medical device; examples are paralysis, Down’s syndrome, or a severe learning disability to avoid bias caused by testing users with unrepresentative conditions
Does not have any broken bones or other physical impairments that would prevent them from using a medical device
Is not a licensed healthcare provider and did not receive formal training beyond CPR/first aid
Does not have experience injecting insulin into him/herself or others
Has not previously used or interacted with the insulin pump
As the focus of this study is to examine the behavior of healthy adults with normal cognition, recruitment of the elderly population will be included in the scope of this study. Adding children as an additional user group would double the sample size since their characteristics, capabilities, and limitations are concluded to be distinct in comparison to adults. As a result, the pediatric population will be excluded from this study.
Participants who frequently use drug delivery products on themselves or others, including nurses, physicians, and caregivers, will be excluded from the study. These more experienced users will likely have a higher level of aptitude, regarding their initial training as well as their training decay, and by limiting the sample to relatively inexperienced laypeople, results will better generalize to the overall population. In addition, the results may potentially serve as a worst-case training decay model for more advanced or experienced users. The study will rely on self-reported answers to screen out ineligible participants, during recruitment.
The screening criteria will be provided to the recruiters to use, during the screening process. If the moderator determines that a participant who attends the test sessions should have been excluded, during recruitment, because he or she does not meet an inclusion criterion or due to safety concerns (for example, because of intoxication), the study moderator will exclude him or her and seek a replacement; the data from the excluded participant will not be included. Any such exclusion shall be documented.
A systematic review of skill retention and skill decay literature with retention intervals between 1 day to 7 days was conducted. In result, 15 articles provided 48 data points and a minimum average effect size of 0.316. Given this effect size, the current participant budget would allow for 4 cohorts of 29 or more participants, each (116 or more in total), to ensure a statistical power greater than 80% with type I error rate of 5%.
The
Cohen’s f
statistics was used to measure the effect size (Cohen, 1988). Some
papers only reported the results of the analysis of variance (ANOVA).
In those cases, we calculated the effect size, using the
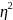 from the ANOVA results. The four proposed retention intervals for
the FDA training decay study are one hour, one day, seven days, and
infinite training decay (no training provided). Effect sizes from
previous studies were collected within this range and then averaged
for each period. If a study tested multiple retention intervals, the
first was taken into consideration to avoid an effect of relearning.
from the ANOVA results. The four proposed retention intervals for
the FDA training decay study are one hour, one day, seven days, and
infinite training decay (no training provided). Effect sizes from
previous studies were collected within this range and then averaged
for each period. If a study tested multiple retention intervals, the
first was taken into consideration to avoid an effect of relearning.
A total of 48 data points were extracted for retention intervals between 1 day and 7 days. The average effect size for each interval are shown in Table 5. The majority of the data were for studies looking at 1 day and 7 days; 54% of data was recorded for 1 day and 27% of data for 7 days. No previous studies were found with a one-hour retention range. The minimum average retention interval effect size was 0.316, and the maximum was 0.654 (Table 5).
Table 5: Average Effect Size by Retention Interval, with the Lowest Average Effect Size Denoted with an Asterisk
Retention Interval (Days) |
Number of Data Points |
Average Effect Size |
1 |
26 |
0.753 |
2 |
5 |
0.610 |
3 |
1 |
2.044 |
5 |
3 |
0.316* |
7 |
13 |
0.826 |
Total |
48 |
|
For the FDA training decay study, the scope has been refined to focus on testing differences in training decay across task types for one medical product. This meta-analysis reviewed previous literature with the inclusion and exclusion criteria stated above to find studies that tested the main effect of task type on retention and determine the sample size required in each retention interval for statistical significance. There were an absence of studies testing task type and memory retention, which demonstrates the need for further research.
With respect to retention intervals, effect sizes were collected and averaged by retention interval to guide the proposed experimental design. To be conservative, the smallest average effect size was selected, i.e., 0.316, to determine the number of retention intervals that could be tested. To guarantee a power of 80% or more, the number of cohorts, minimum cohort sizes, and minimum sample size are listed in Table 6, based on the number of groups desired.
Table 6: Possible Participant Cohort Sizes with Effect Size of 0.316 and Statistical Power of 0.80 with p=0.05 as Well as the Power Based on Average Effect Sizes for Each Retention Interval in Table 5.
-
Number of Groups
Minimum Number of Participants per Group
Total Number of Participants
Power Based on Effect Size
0.316
0.610
0.753
0.826
2.044
4
29
116
0.811
0.999
1
1
1
5
25
125
0.802
0.999
1
1
1
6
24
144
0.833
0.999
1
1
1
7
21
147
0.814
0.999
1
1
1
8
19
152
0.804
0.999
1
1
1
The scope of the UserWise/SJSU Training Decay grant includes testing an approximate maximum of 120 participants as part of the main human factors study. To guarantee a statistical power of 80% or more with significance level of 0.05, only 4 cohorts will be tested (assuming each cohort has a minimum of 29 participants for a minimum total of 116 participants to complete the study). The cohort sizes to be tested in the pilot study and main study are listed in Table 7 and Table 8, respectively.
Twelve (12) participants will be included in the pilot study. Due to the risk of dropout or non-compliance, especially because of the multiple timepoints, it is anticipated that approximately 20% of recruited participants will not complete the study. For purpose of IRB approval, up to thirty (30) participants will be recruited. Refer to Section 6.3 for additional rationale of sample size and the associated calculations.
Table 7 shows the approximate breakdown of the cohorts. Note that randomization, dropout, non-compliance, or other factors may influence these cohort sizes.
Table 7: Participant Cohort Sizes for the Pilot Study
Cohort |
Training Decay |
Minimum Number of Participants per Cohort |
1 |
No Training, No Decay |
N = 3 |
2 |
Training Decay = 1 hour (1h +9m/-0m) |
N = 3 |
3 |
Training Decay = 1 day (24h +/- 3.6h) |
N = 3 |
4 |
Training Decay = 7 days (168h +/- 25.2h) |
N = 3 |
Minimum Total Number of Participants: |
N = 12 |
|
A minimum of 116 participants will be included in the main study. Due to the risk of dropout or non-compliance, especially because of the multiple timepoints, it is anticipated that approximately 20% of recruited participants will not complete the study. For purpose of IRB approval, up to 200 participants will be recruited. Refer to Section 6.3 for additional rationale of sample size and the associated calculations.
Using their experience recruiting non-college student populations, San Jose State University will make an effort to recruit, such that our sample population mirrors the U.S. adult population.
Table 8: Participant Cohort Sizes for the Main Study Based on the Conservative Effect Size of 0.316 with a Statistical Power of 0.8 with p=0.05.
Cohort |
Training Decay |
Minimum Number of Participants per Cohort |
1 |
No Training, No Decay |
N = 29 |
2 |
Training Decay = 1 hour (1h +9m/-0m) |
N = 29 |
3 |
Training Decay = 1 day (24h +/- 3.6h) |
N = 29 |
4 |
Training Decay = 7 days (168h +/- 25.2h) |
N = 29 |
Minimum Total Number of Participants: |
N = 116 |
|
To address the goal of quantifying training decay curves, participants will be randomly assigned via a block-randomization scheme into one (1) of four (4) cohorts, specifying the length of time before they return for a follow-up assessment and the type of device they will be evaluating.
One important part of this study is ensuring the participant cohorts do not differ in any meaningful way so that any detected differences can be attributed solely to their cohort assignment and not to pre-existing differences. By randomly assigning the participants into the various cohorts, they should be matched on demographics, such as age, sex, education, and income. For all potential variables, the appropriate statistical test will be performed (t-tests for continuous variables, chi-squared for categorical variables) to ensure a lack of significant differences between the participant cohorts.
Participants will receive a token of appreciation. The payments were calculated, using current industry best practice, and are kept as low as reasonably possible in order to avoid undue pressure to participate:
Participants may be invited to participate in up to two (2) study sessions, depending on the cohort to which they are assigned.
Participants may earn up to one hundred and fifteen dollars ($115) if they participate in both sessions (approximately 2.5 hours, total). Payments to participants are as follows:
$75 for completing one session (first session: approximately 90 minutes)
$40 for completing a second session at a specific later date (second session: approximately 60 minutes).
SJSU students may opt to receive course credit that approximately matches the length of the session(s) completed instead of receiving the incentive amount.
Participants are expected to complete a total of 2.5 hours in the human factors study session(s). If the participant is scheduled in a cohort with no training, he or she will only be asked to participate in one (1) session, lasting approximately 90 minutes. For participants who are scheduled to participate in two human factors study sessions, the first session is expected to last 90 minutes, and the second session is expected to last 60 minutes, for a total of approximately 2.5 hours, combined. These estimates are based on the extensive experience of the researchers.
Participants will not be reimbursed for travel or accommodations.
In line with FDA guidance and best practice, the evaluation sessions will be conducted in a way that simulates expected use. Participants will be given an opportunity to use the device independently and without guidance, coaching, or intervention from the Study Moderator.
The Moderator will introduce each task, describe the nature of the use scenario in general terms, and guide the participant from one scenario to the next, when appropriate. The moderator will use a workbook/script to ensure consistency across all participants. Typically, during each evaluation session, the Moderator will intervene only to avert potential harm to the participant.
Each study session will include the following subsection, as noted:
Introduction and Consent
Participant Training (omitted for Cohort 1)
Baseline Assessment
Training Decay (as designated per cohort in Table 7 and Table 8, omitted for Cohort 1)
Main Assessment (omitted for Cohort 1)
End of Session Questionnaire
Prior to the start of the participant’s training, the participant will be provided with the Informed Consent Form. Once the participant has read and signed the Informed Consent Form, initial introduction and background to the study will be provided before the training (or study session for the cohort with no training). See Section 8.1 for additional information, regarding the Informed Consent Form.
Participant Training
Participants who are in trained cohorts (as designated per cohort in Table 7 and Table 8) will be provided with training relevant to the insulin pump. The training will include videos, which reference the User Manual, and a hands-on component. Participants will have the opportunity to ask a trained Moderator clarifying questions at the end of the training.
The training will be scheduled to take approximately 30 minutes. The overall length of this training session as well as the presence or absence of clarifying questions and the questions, themselves, will be recorded.
Discussion of Confounding Variables for Training & Study Limitations
Training length is not believed to be a significant confounding variable for the purpose of this study since all participants will have training sessions that are similar in length (approximately 30 minutes). It is not within the scope of this study to examine the use of extremely complex medical devices requiring more than 1 hour of training. Further, medical devices used by lay users commonly require shorter training lengths (e.g., close to 1 hour) than training offered to healthcare providers (e.g., 3-day training for an interventional surgical device), due to limitations of patient time and healthcare provider time (in the case that the healthcare provider is giving the training).
Training length is not believed to influence training decay significantly as long as each participant covers the same materials. Some participants may have higher aptitudes, leading to shorter training lengths, while other participants may require the full amount of time allotted to receive all of the training. Overall, all participants in cohorts with training will be offered identical training content, apart from the final portion of the training in which the participant will be allowed to ask questions (which is what would likely occur in real life). All participants in cohorts with training will be trained by means of a training video, which will provide an overview of the insulin pump and a walkthrough demonstration of the setup and operation. The training video will cover all tasks listed in Table 10. In case participants ask questions after the training, the Moderator or Observer will record the question and find the relevant section of the training video script or User Manual. The Moderator or Observer will read the relevant section in the training video script or User Manual aloud to the participant. If the information required to answer the participant’s question is related to the tasks evaluated in the study session but not in the training video or in the User Manual, the Moderator or Observer will defer the question until the completion of the participant’s second study session. If the question is related to the Informed Consent Form, participant safety, or another aspect of the study, which would not affect task performance or otherwise bias the participant, the Moderator or Observer will answer the participant’s question.
Literature indicates that depth of learning (i.e., quality of training) can impact training decay. The intent of this study is to evaluate retention of information, given that a user achieves a “good” depth of learning. In order to limit the impact of variability in training quality, the team will make a concerted effort to generate training that is of good quality. The training quality will be initially evaluated in the pilot study, where a cohort with no training will be included, which will allow for a qualitative assessment of participant performance without training and immediately after training to inform the depth of learning.
The method of training (e.g., hands-on vs. self-training, through reading) can influence the depth of learning. In an effort to keep the training consistent between participants, the training employed during this study is limited in scope to a walkthrough of the instructions by means of a training video, with participants’ having the opportunity to follow the video along with the product.
In addition, training decay could be influenced by training repetition. Since medical device training is not commonly repeated in the field, this attribute will not be explored.
This study will measure participant performance on each task, during training and after training decay, to calculate training decay curves. Evaluation of device efficacy is out of scope for this human factors study. As such, this study is not assessing whether the labeling of this device is safe and effective; thus, the product User Manual will not be evaluated through findability or comprehension questions.
Immediately after the training session, participants who are in trained cohorts will undergo a baseline human factors test to assess their performance with no training decay. The baseline assessment will include the exact same Simulated Use and Knowledge Task questions that will be used for the main human factors study session (see Section 7.1.5). This baseline assessment is not applicable to participants in a cohort without training, as those participants will only participate in one study session, described further in Section 7.
Task success will be collected for each task as outlined in the Moderator’s Script and/or Data Sheet, which will allow the study team to assess participant performance at timepoint zero. This assessment will be used as a baseline assessment with which future timepoints can be compared. The training decay time will begin once they conclude their baseline assessment.
Each participant will be trained on and asked to use the insulin pump to determine the training decay curves for various task types.
Scenario test cases with expected responses are detailed in Table 10.
It is expected that the User Manual will be available with the purchase of the insulin pump. To represent the actual use environment, study participants will have the User Manual available for reference should they choose to use it, during the Simulated Use and Knowledge Task questions.
Instances of referring to the User Manual, during the study, will be recorded.
The Moderator may ask the participant to find and interpret information in the User Manual at the end of the session if deemed necessary, such as in cases when:
A participant is unsuccessful in a Critical Task
The Use Error is mitigated with the User Manual
Knowledge Tasks
After completing the Simulated Use portion of the study, the Moderator will ask follow-up questions to assess the participant’s knowledge of how to perform the tasks related to use of the device.
In order to prevent reinforcement of the training prior to the second human factors study session, the Moderator and/or Observer will not ask Subjective Feedback questions or probe for root cause for any tasks, during the baseline session, including those rated as Difficulty or Unsuccessful. However, the Moderator will look at possible root causes, based on observation of preceding events or unprompted comments from the user. In any scenario in which the participant seems to be “guessing” about what he or she needs to do next, during simulated use or the knowledge tasks, the Moderator will make a note of the confusion. The Moderator will not follow up with questions to identify the root cause until the end of the second study session, as appropriate.
Using a block-randomization scheme, participants will be randomly assigned to a cohort to determine the length of training decay they will experience. Depending on their cohort assignment, participants will be assigned one of the following training decay periods:
Cohort 1 - No Training (and no training decay)
Cohort 2 - Training Decay of 1 hour (1 hour +9/-0 minutes),
Cohort 3 - Training Decay of 1 day (24 hours +/- 3.6 hours),
Cohort 4 - Training Decay of 7 days (168 hours +/- 25.2 hours).
Training decay is not applicable to participants in a cohort without training (Cohort 1), as those participants will only participate in one study session, described further in Section 7.
As specified in the parentheses, some imprecision, regarding exactly when they return for the second session, for the main assessment will be allowed for the sake of practicality. It is unrealistic to have no allowance in the training decay time, due to difficulties in scheduling participants. Additionally, having no training decay time allowance would lead to a higher rate of data to be unusable, due to no shows’, late participants’, or unforeseen events’ delaying the start of the second study session. This study allows for up to a 15% variance from the target training decay period, except for the one-hour decay cohort, where the training decay length will not be permitted to be less than one hour. The actual time of decay will be recorded for each participant. Participants will be asked to use the device, again, with the same task success collected to assess which steps they are able to complete correctly.
Participant Activities Between Sessions
During the training decay period between the study sessions, participants will be doing a variety of activities in their personal lives with variable and unpredictable amounts of distraction. This can lead to accelerated decay if their activities distract them to an extent that interferes with their training retention. No restrictions will be imposed on participants’ actions, as such restrictions would make the decay period more unnatural. However, the study personnel will ask questions at the follow-up assessment to capture what participants thought about the training and how stressed and fatigued they were feeling.
The cohort asked to return in one (1) hour is unique in that it will not be going home in between sessions. The members of this cohort will be asked to wait in a waiting room in the same building and will be allowed to do whatever they like, aside from practicing use of the device. The study personnel will ask questions at the follow-up assessment about their activities, during the one-hour break. The feedback on their activities between sessions will be used for informational purposes, only, as it is not within scope to assess accelerated decay to a degree to which it would be statistically significant.
Simulated Use
Participants will be provided with the same scenario, study articles, equipment, and prompt as the Baseline Session. The Simulated Use portion of the study, as described in Section 7.1.3.1, will be repeated for the main assessment.
The Moderator will ask follow-up questions to assess the participant’s knowledge of how to perform the tasks, related to use of the device.
In addition, the participant will be probed for root cause (after completing his or her final use of the product) to determine if he or she was unsuccessful, had difficulty, or did not perform critical tasks, during the simulated use portion of the study. In any scenario in which the participant seems to be “guessing” about what he or she needs to do next, during simulated use or the knowledge tasks, the Moderator will make a note of the confusion and follow up with questions to identify the root cause.
Following a completion of the simulated use and knowledge assessment portions of the study, subjective data will be collected to better explain the findings. In order to prevent reinforcement of the training, prior to the second human factors study session, the Moderator and/or Observer will not ask Subjective Feedback questions, during the Baseline Assessment of participants, returning for a second session. This will include a subjective assessment of any use difficulties experienced during the test.
The participants may be asked questions about their experience using the device, such as:
“Were there any points, during the study, at which you experienced difficulty or had concerns about the device or its labeling?”
“Did you experience any moments of hesitation, while using the device?”
“Do you have any safety concerns, regarding this device or its labeling?”
In addition, information about the user’s experience will be collected. Confounding variables can arise, simply due to unpredictable circumstances on the day of assessment. Participants will also be asked about their activities between timepoints, when appropriate, such as if they thought about their training, what details of the training they thought about, how stressed they were feeling, and how tired/fatigued they were feeling, during the session(s). All of these variables may affect their ability to perform the assessment and will be a useful record for informational purposes and secondary assessment.
Participants will be asked to complete a questionnaire at the end of their final human factors evaluation session. The questionnaire will contain three key sections: 1) the Newest Vital Sign (NVS) test; 2) task difficulty ratings; and 3) demographics questions, described further in the sections below.
The Newest Vital Sign (NVS) Test
Although the study is excluding participants with extensive experience using medical devices, it is expected that many participants will have at least some prior experience using similar devices. After the completion of the participant’s last study session, each participant will be assessed for health literacy levels, using the Newest Vital Sign (NVP) assessment, a standardized assessment of health literacy (Weiss, 2005). As with other confounding variables, the data will be assessed and tested for differences between cohorts and included in the Study report if deemed necessary.
One important additional analysis is examining the relationship between task difficulty and its training decay curve. It is hypothesized that the difficulty of the task is likely to influence memory retention, significantly. To analyze this variable, we will ask participants to assess the difficulty of tasks, using a Likert scale after completing all simulated use portions of the study. This data collected will be analyzed statistically, using the methods described in Section 7.7.1.
Demographics Questions
Demographics will be collected at the end of the session in order to minimize potential bias within the study sessions. These demographics are being collected in order to analyze performance in relation to characteristics such as age, sex, ethnicity, education, and profession, and to inform whether there are significant correlations between variables.
It is hypothesized that the training decay associated with different tasks types (i.e., cognitive, psychomotor/motor, or perceptual) may have different training decay curve profiles (Clark, 2016). In other words, tasks requiring cognitive skills may have different training decay curve profiles than those of tasks requiring psychomotor/motor skills or perceptual skills and vice versa.
To test this hypothesis, tasks will be categorized as either a cognitive, psychomotor/motor, or perceptual task. Each type of task will be tested in the simulated use portion of the study sessions. A literature review was conducted to compile a definition of each of the task types.
A systematic literature review is being conducted with an initial finding of 25 articles related to task categorization and task taxonomy. All articles were evaluated by SJSU undergraduate students for relevancy. Seven articles were read in-depth, with mention of 16 existing taxonomies for this preliminary evaluation. The authors found overlap between these taxonomies and summarized task types in the following broad categories:
Cognitive, psychomotor, and perceptual (Hancock, Ross, & Szalma, 2007)
Cognitive, motor, and perceptual-motor (Meador & Hill, 2011)
Cognitive, psychomotor, and interpersonal (Arthur, Bennett, Edens, & Bell, 2003)
Based on the literature review, the following definitions for the three task types will be used to categorize tasks for the purpose of this research:
Cognitive Tasks: Tasks that require a person to mentally process new information (i.e., acquire and organize knowledge/learn) or allow them to recall and retrieve that information from memory or to use that information at a later time in the same or similar situation (i.e., transfer) (Kirschner, 2012). Cognitive tasks are related to the thinking, idea generation, understanding, problem-solving, or the knowledge requirements of the job (Arthur, Bennett, Edens, & Bell, 2003). Cognitive tasks include performing a mathematical calculation, processing information mentally, and reaching conclusions (Meador & Hill, 2008). An example could be problem solving to figure out how much money you could withdraw from an ATM, while still maintaining a minimum balance.
Psychomotor/Motor Tasks: Tasks that include physical activities which involve a range of movement from very fine to gross motor coordination (Arthur, Bennett, Edens, & Bell, 2003). An example could be removing cash from the machine.
Perceptual Tasks: Tasks that require the person to identify or separate targets or objects (Meador & Hill, 2008) and respond to those targets (Mead & Fisk, 1997). An example of this task could be seeing a cue to enter a pin code.
As this literature review is still ongoing, the task categorization listed above may change, prior to the start of the main study.
Tasks have been independently categorized, based on these definitions, by two experienced research team members and adjudicated with a third team member with intimate experience of the device and tasks. When selecting the task type, the researchers selected the type that is expected to be the primary contributor to workload and not the critical use error type. If a task had two or more equal contributors to the workload, the task was subdivided into multiple tasks. The task types for tasks being tested in this research are noted in Table 10.

Figure 7: Task Type Categories
A task analysis (in the form of a workflow, shown in Figure 6) and a use-related risk analysis were developed to identify the tasks associated with using the insulin pump. The Critical Tasks associated with the patient profile will be evaluated in this human factors study.
As stated in the FDA’s 2016 Guidance for Human Factors (U.S. Food and Drug Administration (FDA), 2016) (2016 Guidance), Critical Tasks should be identified and evaluated, during an HF validation Study. Following the FDA’s 2016 Guidance, the Task Analysis was a principal input for the Use-Related Risk Analysis (UserWise Inc., Document Number 066), which involved making a Perception, Cognition, Action (PCA) error analysis that identified tasks associated with severe consequences.
In the Use-Related Risk Analysis, consequence severity was rated conservatively, with the worst foreseeable consequences identified for each hazard-related use scenario. The severity ratings used are shown in Table 9.
Table 9: Task Severity Ratings from the Use-Related Risk Analysis Used to Categorize Task Severity
Severity Level |
Definition |
S5 - Catastrophic |
Results in death |
S4 - Critical |
Results in permanent impairment of a body function or permanent damage to a body structure; life-threatening injury |
S3 - Serious |
Results in injury or impairment requiring professional medical intervention to prevent:
(excludes trivial impairment or damage) |
S2 - Minor |
Resulting in temporary injury or impairment not requiring professional medical intervention |
S1 - Negligible |
Results in inconvenience, temporary discomfort, or minor impairment |
S0 – None |
No impact to health or well being |
User tasks associated with risks having a severity rating of 2, 3, 4, or 5 were identified as Critical.
Tasks. Tasks associated with risks having a severity rating of 0 or 1 were identified as Non-Critical Tasks and were not included in Table 10, below.
To replicate FDA’s requirement for medical devices and combination products, this study will focus on evaluating Critical Tasks for data collection.
Scenario test cases and acceptance criteria are detailed in Table 10, below.
Table 10: Critical Tasks and Failure Criteria
# |
High-Level Workflow Task |
Sub-Task |
Task Type |
Participant Response Indicating Critical Use Error |
Risk IDs |
1 |
Basic Programming |
Set the time (Select either the 12-hour set up or 24-hour set up) |
Cognitive |
User inadvertently selects wrong time setup (between 12 hours and 24 hours) and does not correct the time. |
1693 |
2 |
Basic Programming |
Set the time (hours, am / pm) |
Psychomotor/Motor |
User inadvertently sets incorrect time (hours, am/pm) and does not correct the time. |
1897 |
3 |
Basic Programming |
Set the time (minutes) |
Psychomotor/Motor |
User inadvertently sets incorrect time (minutes) greater than 30 minutes2 and does not correct the time. |
1859 |
4 |
Prepare the Vial |
Remove reservoir from the package |
Psychomotor/Motor |
User drops sterile reservoir in such a way that there is potential contamination of the sterile reservoir septum. User proceeds to use potentially contaminated reservoir. |
1174 |
5 |
Prepare the Vial |
Inspect the vial liquid |
Cognitive |
User does not inspect the vial to ensure the liquid is free of particulates, colorless, and clear. |
1327, 1840 |
6 |
Prepare the Vial |
Check vial expiration date |
Cognitive |
User does not check the vial expiration date to ensure liquid is not expired. |
1363 |
7 |
Prepare the Vial |
Remember to swab the vial with alcohol |
Cognitive |
User does not swab vial, or user knowingly contaminates the vial after swabbing and forgets or does not understand the need to clean it with alcohol, afterward. User proceeds to use potentially contaminated vial. |
1188 |
8 |
Prepare the Vial |
Swab the vial with alcohol |
Psychomotor/Motor |
User unknowingly contaminates the vial septum and does not swab it with alcohol, afterward. User proceeds to use potentially contaminated vial. |
1056 |
9 |
Fill the Reservoir
|
Attach transfer guard to the vial with plunger extended |
Psychomotor/Motor |
User places his or her finger inside either end of the transfer guard, such that there is potential for a needle stick with a clean needle.
Note: If this situation occurs, the Moderator shall attempt to intervene and stop the participant as soon as possible. |
1382 |
User disconnects the reservoir and/or transfer guard more than 3 times when attempting to transfer the liquid, and/or the study Moderator observes particulate from the vial septum’s being released into the fluid. |
1060 |
||||
10 |
Fill the Reservoir |
Flip the vial over so that the vial is on top |
Psychomotor/Motor |
User drops vial, transfer guard, and/or reservoir in such a way that there is potential contamination of the sterile needle and/or septum of the vial or reservoir. User proceeds to use potentially contaminated vial, transfer guard, and/or reservoir. |
1919 |
11 |
Fill the Reservoir
|
Slowly pull down on the plunger rod to fill the reservoir
|
Psychomotor/Motor |
User pulls down on the plunger too quickly, causing cavitation in the liquid. User injects fluid with small air bubbles. |
1133 |
User pulls down on the plunger too aggressively and pulls out plunger. Liquid is spilled, and dose is lost. User places plunger back in reservoir and continues to use same (now contaminated) reservoir and plunger. |
1620 |
||||
12 |
Fill the Reservoir
|
Remember to gently tap the side of the reservoir and push the plunger (to remove large air bubbles) |
Cognitive |
User forgets to remove air bubbles from the reservoir.
|
1555, 1748
|
13 |
Fill the Reservoir
|
Gently tap the side of the reservoir and push the plunger (to remove large air bubbles) |
Perceptual |
While tapping, user drops vial, transfer guard, and/or reservoir in such a way that there is potential contamination of the sterile needle and/or septum of the vial or reservoir. User proceeds to use potentially contaminated vial, transfer guard, and/or reservoir. |
1945 |
14 |
Set Up the Reservoir
|
Flip the vial over (upright) and disconnect the reservoir from the vial
|
Psychomotor/Motor |
User does not flip the vial over (upright) and gets fluid on the top of the reservoir and/or infusion tubing set when disconnecting. Fluid temporarily blocks the vents, during dose delivery, and results in an underdose. |
1173, 1101, 1193 |
User drops vial and/or reservoir in such a way that there is potential contamination of the sterile vial or reservoir septum. User proceeds to use potentially contaminated vial and/or reservoir. |
1318 |
||||
15 |
Set Up the Reservoir |
Remember to dispose of the transfer guard in a sharps container |
Cognitive |
User does not dispose of the transfer guard in a sharps container.
Note: If this situation occurs, the Moderator shall ensure proper disposal of the needle in a sharps container at the end of the study session. |
1357 |
16 |
Set Up the Reservoir |
Attach tubing connector onto the reservoir |
Psychomotor/Motor |
User places their finger in tubing connector, such that there is a potential for a needle stick injury to his or her finger with the clean needle.
Note: If this situation occurs, the Moderator shall attempt to intervene and stop the participant as soon as possible. |
1522 |
17 |
Set Up the Reservoir |
Twist to lock connector onto reservoir |
Psychomotor/Motor |
User does not twist to lock connector to reservoir, such that the reservoir has the potential to become detached from the tubing connector. |
1246, 1634, 1905 |
User partially twists to partially lock the connector onto the reservoir, such that the reservoir has the potential to become detached from the tubing connector. |
1697, 1139, 1804 |
||||
18 |
Set Up the Reservoir |
Remember to push up on the plunger until liquid is in the tubing to purge air bubbles from the reservoir |
Cognitive |
User does not push up on the plunger to remove air bubbles. |
1218 |
19 |
Set Up the Reservoir |
Push up on the plunger until liquid is in the tubing to purge air bubbles from the reservoir |
Perceptual |
User has infusion tubing connected to the injection site and pushes the plunger to remove air bubbles but does not see the fluid entering the tube and inadvertently injects fluid from the reservoir through the tubing and into the injection site. |
1463 |
20 |
Prepare the Infusion Set |
Rewind the insulin pump plunger |
Perceptual |
Infusion set is attached to the injection site. Pump prompts user to rewind pump, and user presses ESC instead of ACT and does not rewind the pump, prior to inserting the reservoir. Fluid is pushed out of the reservoir and into the infusion set tubing. |
1958, 1389, 1285 |
21 |
Prepare the Infusion Set
|
Insert the reservoir into pump case |
Psychomotor/Motor |
User partially turns reservoir, not fully locking reservoir into pump case unit. Reservoir is not fully seated, leading to a missed dose (plunger rod does not contact the rubber reservoir stopper). |
1654 |
User drops the reservoir when attempting to insert into pump case, such that there is potential contamination of or damage to the reservoir and/or infusion set. User proceeds to insert and use reservoir. |
1940, 1233 |
||||
22 |
Prepare the Infusion Set
|
Fill infusion set tubing before connecting to the injection site |
Cognitive |
User has already connected the infusion set to the body, and user inadvertently selects "Yes" to the on-screen prompt, asking if the infusion set is disconnected from the body in the FILL TUBING screen. User proceeds to fill tubing, and liquid enters the injection site. |
1019 |
23 |
Prepare the Infusion Set |
Select the correct response on the FILL TUBING screen |
Cognitive |
User inadvertently presses ESC instead of ACT in the FILL TUBING screen. User proceeds without filling the infusion set. |
1220 |
24 |
Prepare the Infusion Set |
Fill infusion set tubing until liquid is visible |
Perceptual |
User has infusion tubing disconnected from the body but does not press and hold ACT until liquid is visible from the end of the infusion set needle. |
1738 |
25 |
Insert the Infusion Set |
Remember to wash hands or use hand sanitizer |
Cognitive |
User does not wash hands or use hand sanitizer. |
1787 |
26 |
Insert the Infusion Set |
Select the injection site |
Cognitive |
User selects an appropriate area of the body to place the injection pad and perform the injection. Appropriate areas include: stomach (outside of 2 inches around the navel), upper thighs, back of arm, lower back, and buttocks. |
1948 |
User indicates he or she would apply the infusion set to inject into a blood vein. |
1283 |
||||
User does not understand the need to rotate the injection site. |
1672 |
||||
27 |
Insert the Infusion Set |
Remember to clean the injection site (on the injection pad) with alcohol |
Cognitive |
User does not clean the injection site (on the injection pad) with an alcohol swab or contaminates the injection site after cleaning it. |
1716 |
28 |
Insert the Infusion Set |
Remove both adhesive cover strips from the infusion set |
Psychomotor/Motor |
User removes only one adhesive strip and not both, resulting in the infusion set’s not securely attaching to the skin (this could lead to the potential of the cannula’s partially dislodging). |
1809 |
29 |
Insert the Infusion Set |
Safely remove the needle guard |
Psychomotor/Motor |
User removes the needle guard, such that there is potential for a needle stick injury to his or her finger with the clean needle.
Note: If this situation occurs, the Moderator shall attempt to intervene and stop the participant as soon as possible. |
1501 |
30 |
Insert
the |
Apply infusion set to selected injection site
|
Psychomotor/Motor |
User applies the infusion set outside of the cleaned injection site. |
1099 |
User does not press around the adhesive, and the infusion set is not sealed to the injection site (this could lead to the potential of the cannula’s partially dislodging). |
1033 |
||||
User presses around some of the adhesive and partially seals infusion set onto the injection site (this could lead to the potential of the cannula’s partially dislodging). |
1901 |
||||
31 |
Insert the Infusion Set |
Remove the needle housing from the infusion set |
Psychomotor/Motor |
User removes the exposed quick set needle, such that there is a potential for a needle stick injury.
Note: If this situation occurs, the Moderator shall attempt to intervene and stop the participant as soon as possible. |
1952 |
32 |
Insert the Infusion Set |
Place the needle guard over the needle |
Psychomotor/Motor |
User places the needle guard over the quick set needle or otherwise handles the exposed quick set needle, such that there is a potential for a needle stick injury.
Note: If this situation occurs, the Moderator shall attempt to intervene and stop the participant as soon as possible. |
1443 |
33 |
Insert the Infusion Set |
Remember to dispose of the needle housing in a sharps container |
Cognitive |
User does not dispose of the contaminated infusion set needle housing in a sharps container.
Note: If this situation occurs, the Moderator shall ensure proper disposal of the needle in a sharps container at the end of the study session. |
1222 |
34 |
Insert the Infusion Set
|
Remember to fill the cannula 0.5 units |
Cognitive |
User forgets the expected fill volume is 0.5 units and enters an incorrect amount. |
1288 |
User fills the cannula to 0.5 units |
Perceptual |
User intends to fill to 0.5 units but misunderstands on-screen prompt and/or inadvertently enters an incorrect amount. |
1306 |
||
User inadvertently presses ESC to skip the fill cannula step. |
1680 |
||||
35 |
Basal Dose |
Navigate to the Basal menu |
Cognitive |
User inadvertently selects bolus, temp basal, or other setting, while attempting to set basal dose. |
1646 |
36 |
Basal Dose |
Recognize that the first basal time default is midnight |
Perceptual |
User does not see or understand that the start time for first basal dose is at midnight (shown on the screen). User sets an additional basal dose over what is anticipated. |
1882 |
37 |
Basal Dose |
Enter basal rate and time |
Cognitive |
User
enters incorrect basal dose amount. May occur due to: |
1383 |
User enters incorrect start time for the basal dose, differing by more than 30 minutes from the intended start time. |
1332 |
||||
User inadvertently enters additional times and rate(s), beyond what is needed (up to 3 additional doses). |
1794 |
||||
38 |
Bolus Dose |
Navigate to the Bolus menu |
Cognitive |
User inadvertently selects Basal, while attempting to set Bolus, and sets the times, such that the dose is not immediately delivered, leading to a delay in delivery. |
1674 |
39 |
Bolus Dose |
(If Dual/Square Wave is on) Select Normal Bolus |
Cognitive |
Dual/Square option is on, and user inadvertently selects the incorrect bolus type (does not select normal bolus). User receives correct amount of bolus at a different rate/time. |
1942 |
40 |
Bolus Dose |
Input desired bolus amount |
Cognitive |
User inadvertently inputs incorrect amount for the bolus. |
1205, 1319, 1195 |
User inputs correct amount of bolus but unknowingly cancels the bolus dose instead of pressing ACT. User believes the bolus dose is delivered, when it is not. |
1452 |
||||
41 |
Change the Infusion Set |
Knows to remove the infusion set from the injection site |
Cognitive |
User indicates he or she would leave the same infusion set attached on the injection site for more than three days. |
1455 |
42 |
Change the Infusion Set |
Knows to remove the reservoir from the infusion pump |
Psychomotor/Motor |
User does not remove the reservoir from the pump to reuse it for the next infusion set. |
1233 |
43 |
Change the Infusion Set |
Remembers to dispose of the used infusion set in a sharps container |
Cognitive |
User does not dispose of the infusion set in a sharps container.
Note: If this situation occurs, the Moderator shall attempt to intervene and stop the participant as soon as possible. |
1661 |
44 |
Replace the Battery |
Tighten battery cap, so the slot is aligned horizontally with the pump |
Psychomotor/Motor |
User does not fully tighten the battery cap, such that the battery cap has the potential to come loose over time, leading to the pump’s turning off. |
1782, 1608, 1006 |
During each study session, participants will perform tasks independently and in a natural manner, without guidance, coaching, praise, or critique from the study personnel.
Observer(s) and Moderator(s) will observe for use errors. Data on participants will be collected as thoroughly as possible to ensure the necessary information is gathered to isolate the targeted effects.
For the main study session, the Moderator will also ask probing questions to reveal any use errors that were not conveyed through observation. As time permits, performance failures will be followed with questions to identify how and why the participant believes the failure occurred. The Moderator will not ask probing questions for the baseline assessment, as described in Section 7.1.3.
Participants will not be allowed a “second chance” to perform a task correctly after a failure. That is, if an irreversible safety issue is observed in which a user would not normally have a “second chance” to resolve the issue, then any subsequent attempts to complete the task will serve as input only for the purpose of continuous human factors-improvement.
The Observer(s) and/or Moderator(s) will note any instances in which users have trouble finding information and cases in which they refer to the incorrect information. Instances of moderator assistance will be recorded.
In any scenario in which the participant seems to be “guessing” about what he or she needs to do next, the Study Moderator will make a note of the confusion in the data sheet and indicate the appropriate section of the User Manual to the participant. Final determination of task success will be recorded.
The purpose of this study is not to evaluate the safety or effectiveness of a medical product but, instead, to explore the various effects of training decay. Thus, the study will not follow strict Good Documentation Practice (GDP) guidelines, using a paper-based entry. Instead, computer-based entry will be performed, using data sheets; paper-based entry may be performed although this will also not follow strict GDP. The data sheets will be used to facilitate note-taking and the capture of use errors and difficulties that are observed, during each session. Information noted in the Data Sheet will be captured for each participant enrolled in the study.
The Study Moderator and/or Study Observer will review and verify each completed Data Sheet. The document number associated with the Data Sheets will be captured in the Study report, along with the revision letter(s) used during the study.
The Data Sheets will include the following data required to be captured, during each session:
Participant ID
Performance outcomes for each task tested
Subjective assessment data
Root-cause analysis data
In the case that a Use Error is observed, the Study Moderator will perform a root cause analysis, as time allows. Both objective data and subjective data will facilitate identification and understanding of the root causes of use failures or problems that occur.
Photo, audio, and video recordings will be implemented, during the study, to facilitate post-study analysis of use errors and difficulties. Video will only be reviewed in cases where a task needs to be evaluated further or if insufficient data was collected in the data sheets. No media recordings of participants will be shared outside of what is necessary, following the approved IRB application.
Each Critical Task will be attributed to one of the following ratings:
Successful: User is able to complete the task safely and effectively.
Success with no issues (S): User is able to complete the task safely and effectively without Use Errors or difficulties.
Difficulty (D): User is able to complete the task safely and effectively but had significant hesitation or challenges, while completing the task, or user has difficulty or makes a User Error, but the user takes an action to recover and prevents the harm from occurring (i.e., near-miss).
Unsuccessful (U): User is unable to complete the task or committed a use error and is unable to recover.
Did Not Perform or Not Assessed (DNP or NA): User is unable to perform the task, due to: 1) a previous use error that rendered it impossible to perform the task; or 2) the session’s running out of time. Performances that scored as Did Not Perform or Not Assessed were removed from the overall task performance totals and will be summarized separately in the report.
Did Not Record (DNR): The note-taker did not observe the activity, due to proximity or location, or was not able to make an assessment at the time of the observation. Any task marked as Did Not Record will be followed up as necessary, with a review of data captured by the Observer and Moderator after the session and/or video review to determine the appropriate rating.
Difficulties will not constitute task failure in the context of the study. In the case that the participant notes that he or she would not proceed and would use a back-up device, depending on the task, the Moderator/Observer will either provide a back-up device, if available, or make a note of the request in the datasheet and ask the participant to proceed, using the existing device for the purpose of the study. If the participant requests a back-up device, it will not necessarily constitute a “task failure,” but instances will be recorded and evaluated in context.
If a participant indicates he or she would call a customer service number, during simulated use, the participant will be prompted to find the number in the user documentation. After the participant locates the phone number, he or she will be handed a cell phone with “customer service” or another appropriate identifier programmed into the phone. The Study Moderator, Observer, or Facilitator will receive the phone call and simulate a customer-service call. The Moderator/Observer/Facilitator will direct the participant to relevant sections of the User Manual that answer the participant’s question. Instances of calling customer service will be recorded but do not necessarily constitute a task failure.
Any instance of Moderator assistance will be noted and assessed, based on the extent of the assistance provided. Final determination of task success will be recorded.
The data obtained from the study will be evaluated and detailed in the study report and will include performance outcomes, statistical analysis, and subjective assessment data.
The purpose of this study is not to evaluate the safety or effectiveness of a medical product or a thorough root cause analysis but is, instead, to explore the various effects of training decay. Thus, risks with patterns of difficulties or unsuccessful performance will not be evaluated further in relation to the risk analysis to determine if further risk mitigation is required.
Due to the moral hazard that exists in the data analysis’ being performed by the data collector, data analysis will be blinded from the data collection process.
The primary goal of the analysis is to measure the training decay; the secondary goal is to examine differences among task types.
To achieve the first goal, we will at first perform an ANOVA to test the difference among the proportion of successes per training decay period. For the purpose of data analysis, either a rating of “successful” or “difficulty” is considered to be success on the task.
Data will be entered in a linear mixed effects model, an extension of the regular linear model that allows for a nested structure in the data. For this study, the two nested grouping levels will be participant and timepoint.
Each participant will have one or two observations; all but one of the cohorts will have one for their baseline assessment, and all cohorts will have one for their follow-up assessment. As such, random intercepts will be specified for the participant grouping variable. This approach leads to a more powerful design as all other effects are examined only after accounting for the within-participant variance.
Timepoint will be entered as a binary variable (baseline vs. follow-up) as well as decay length (e.g., one hour vs. one day) and task type as categorical variables.
If deemed appropriate, other collected data, such as demographics or NVS, will be entered as fixed effects as well. The inclusion of other parameters, such as random slopes, will be determined by assessing if a model with these parameters significantly outperforms a model without them, using tests, such as the likelihood ratio test.
The dependent variable of interest can be quantified as a percentage of task success rate per individual. The statistical model will allow us not only to detect significant differences but to arrive at an estimate of those differences. For example, this approach will allow us to quantify how much decay occurs between the first and second timepoint or how much more decay there is for one task type vs. another.
The frequency of “successful,” “difficulty,” or “unsuccessful” per each task will be also reported for homogeneity, with the usual reporting of medical device human factors studies.
Testing a two-way interaction between timepoint and decay length will address our first goal of quantifying decay curves. It is hypothesized that this interaction will be significant, as the effect of the timepoint variable (i.e., the training decay between their baseline and follow-up timepoints) will be different, depending on the length of their decay. Planned pairwise comparisons between timepoint groups will allow us to characterize the decay curve. A Bonferroni correction will be used to account for multiple testing error. Statistically significant differences between adjacent timepoint groups (i.e., one hour vs. three days) will indicate that significant decay has occurred. Failure to reach statistical significance can mean different things in different contexts. A lack of significance between the baseline group and the one-hour group may indicate that training decay has not occurred yet, while a lack of significance between groups (e.g., three days vs. one week or vs. no training) may be interpreted as a “bottoming-out” of the training decay curve.
To test for differences among task types, the secondary goal, we will test a three-way interaction between the timepoint, decay length, and task type. The previously mentioned two-way interaction between timepoint and decay length represents the training decay curve. An additional interaction between this curve and the task type is expected; that is, the relationship between decay length and timepoint differences depends on the type of task. The between-group differences will be examined in the study report, with a comparison, looking at the differences between task types at each timepoint. The unique training curve for each task type will be quantified by performing the same pairwise comparisons between timepoint groups and each task. Resulting data will be assessed to determine if there are correlations between tasks that are similar in nature (exact task categorization will be based on a complete literature review).
In the end of study questionnaire, participants will be asked to rate the difficulty of tasks on a scale between 1 and 5. Descriptive statistics will be used to determine the tasks that were considered most difficult, and themes between these tasks may be identified by the research team. To measure the impact of difficulty at the individual level, the median difficulty per task type will be used as a covariate in the linear regression model.
All participants will be asked to read and sign the Informed Consent Form, prior to the start of the human factors study session. The Study Facilitator will ensure the Informed Consent Form is completed, prior to the start of the participant’s first study session. The Study Moderator, Observer, or Facilitator will sign the Informed Consent Form after the participant has signed.
The study will comply with 32 CFR 219, 10 U.S.C. 980, and as applicable, 21 CFR Parts 11, 50, 54, 56, 312) (45 CFR Part 46) as well as the ICH and other applicable federal and state regulations.
The participant screener is written to ensure that women, members of minority groups, and the elderly are not excluded from the population tested. The scope of this research is limited to testing cognition of healthy adults as the intended user group. The pediatric population is excluded from this study, as children would be considered an additional user group since their characteristics are concluded to be distinct in comparison to adults, which would, in turn, double the sample size.
It should be noted that this study is not a clinical trial, and users will simulate the use of a medical product without actually administering therapy to themselves or others.
All study personnel interacting with human subjects as part of this research will have completed training on the protection of human subjects.
Only participants who meet the inclusion criteria will be entered into the human factors study as described in Section 6.2.1.
Additionally, the following features of the study are intended to minimize any ethical considerations:
No minors, prisoners, pregnant women, or incompetent adults will be included.
An informed consent process will be in place.
The study personnel are suitably qualified and experienced for the type of study proposed.
Participants’ identities will be protected and will be stored securely.
The risks of harm have been minimized and will be explained to participants as part of the informed consent procedure.
The study personnel are suitably experienced and comply with industry best practice.
The incentives for participants are set at a modest level to minimize any undue pressure to participate.
The outputs from the study, containing identifying information, will not be published, and participant identities will remain secure to the extent permitted by law.
Participants will have access to drinking water, their medications (if required), and breaks as required.
The study venues have first aid kits available and access to call 9-1-1 in the case of an emergency
The study protocol will be submitted for review by the San Jose State University (SJSU) Humans Subjects Research IRB. Testing will only start with approval from the SJSU IRB.
HIPAA is not relevant to this research study. At no stage during the recruitment process will the recruiters seek to gain access to any medical records for any participant.
Personal details of each participant (i.e., name, email, and/or address) will be recorded solely for the purpose of giving informed consent and collecting initial screening information, but from the point at which the training is initiated, a numbering system will be used to protect the identity of the participant and to remove the link between personal details and test data.
The study personnel will send appropriate safety notifications to regulatory authorities in accordance with applicable laws and regulations.
The study personnel must comply with any applicable institution-specific requirements related to the reporting of adverse events (AEs) involving his/her subjects to the IRB that approved this study.
In accordance with ICH/GCP guidelines, the study personnel will inform the Investigator of “findings that could adversely affect the safety of subjects, impact the conduct of this study or alter the IRB’s approval/favorable opinion to continue this study.”
The study personnel will be responsible for promptly notifying the concerned local or institutional IRB of any safety reports and of filing copies of all related correspondence in the study file.
1Definition from IEC 62366-1
2For the purpose of this study, an earlier or delayed dose that is still within 30 minutes of the intended dose delivery will be considered acceptable.
| File Type | application/vnd.openxmlformats-officedocument.wordprocessingml.document |
| File Modified | 0000-00-00 |
| File Created | 0000-00-00 |
© 2026 OMB.report | Privacy Policy