De Novo request under 21 U.S.C. 513(f)(2)(ii)
Medical Device De Novo Classification Process
NEW_Guidance_8-2017
De Novo request under 21 U.S.C. 513(f)(2)(ii)
OMB: 0910-0844
De Novo Classification Process (Evaluation of Automatic Class III Designation)
Guidance for Industry and Food and Drug Administration Staff
Document issued on [insert publication date of FR Notice].
The draft of this document was issued on August 14, 2014.
This document supersedes “New Section 513(f)(2) - Evaluation of Automatic Class III Designation, Guidance for Industry and CDRH Staff” dated February 19, 1998.

An agency may not conduct or sponsor, and a person is not required to respond to, a collection of information unless it displays a currently valid OMB control number. The OMB control number for this information collection is 0910-xxxx (expires xx-xx-xxxx).
See additional PRA statement in Section 5 of the guidance.
For questions about this document regarding CDRH-regulated devices, contact the Division of Industry and Consumer Education (DICE) at 1-800-638-2041, 301-796-7100, or DICE@fda.hhs.gov.
For questions about this document regarding CBER-regulated devices, contact the Office of Communication, Outreach, and Development (OCOD) at 1-800-835-4709 or 240-402-8010.
U .S.
Department of Health and Human Services
.S.
Department of Health and Human Services
Food and Drug Administration
Center for Devices and Radiological Health
Center for Biologics Evaluation and Research
Preface
Public Comment
You may submit electronic comments and suggestions at any time for Agency consideration to http://www.regulations.gov. Submit written comments to the Dockets Management Staff, Food and Drug Administration, 5630 Fishers Lane, Room 1061, (HFA-305), Rockville, MD 20852. Identify all comments with the docket number [FDA-2011-D-0689]. Comments may not be acted upon by the Agency until the document is next revised or updated.
Additional Copies
CDRH
Additional copies are available from the Internet. You may also send an e-mail request to CDRH-Guidance@fda.hhs.gov to receive an electronic copy of the guidance. Please use the document number (1760) to identify the guidance you are requesting.
CBER
Additional copies are available from the Center for Biologics Evaluation and Research (CBER) by written request, Office of Communication, Outreach, and Development (OCOD), 10903 New Hampshire Ave., WO71, Room 3128, Silver Spring, MD 20903, or by calling 1-800-835-4709 or 240-402-8010, by email, ocod@fda.hhs.gov, or from the Internet at
De Novo Classification Process (Evaluation of Automatic Class III Designation)
Guidance for Industry and
Food and Drug Administration Staff
This guidance represents the current thinking of the Food and Drug Administration's (FDA or Agency) on this topic. It does not establish any rights for any person and is not binding on FDA or the public. You can use an alternative approach if it satisfies the requirements of the applicable statutes and regulations. To discuss an alternative approach, contact the FDA staff or Office responsible for this guidance as listed on the title page.
Introduction
The purpose of this document is to provide guidance on the process for the submission and review of a De Novo classification request (hereafter a “De Novo request”) under section 513(f)(2) of the Federal Food, Drug, and Cosmetic Act (the FD&C Act), also known as the De Novo classification process. This process provides a pathway to Class I or Class II classification for medical devices for which general controls or general and special controls provide a reasonable assurance of safety and effectiveness, but for which there is no legally marketed predicate device.
FDA’s guidance documents, including this guidance, do not establish legally enforceable responsibilities. Instead, guidances describe the Agency’s current thinking on a topic and should be viewed only as recommendations, unless specific regulatory or statutory requirements are cited. The use of the word should in Agency guidances means that something is suggested or recommended, but not required.
Throughout this guidance document, the terms “we,” “us” and “our” refer to FDA staff from the Center for Devices and Radiological Health (CDRH) or the Center for Biologics Evaluation and Research (CBER) involved in the review and decision-making aspects of the De Novo classification process. “You” and “your” refers to the submitter of a De Novo request and/or related materials.
Background
A device may be classified in class III and be subject to premarket approval (PMA) via several different regulatory vehicles. In accordance with the criteria at section 513(a)(1)(C) of the FD&C Act, FDA may promulgate a regulation classifying, or issue an order reclassifying,1 a device type into class III based on the risks posed by the device and the inability of general and special controls to provide reasonable assurance of the safety and effectiveness of the device. All particular devices of such a type are considered to be in class III and such devices are not eligible for the De Novo classification process.
Alternatively, devices of a new type that FDA has not previously classified based on the criteria at section 513(a)(1) of the FD&C Act are “automatically” or “statutorily” classified into class III by operation of section 513(f)(1) of the FD&C Act, regardless of the level of risk they pose or the ability of general and special controls to assure safety and effectiveness. This is because, by definition, a new type of device would not be within a type that was on the market before the 1976 Medical Device Amendments or that has since been classified into class I or class II. Thus, there would be no available predicate device.
This second scenario is what Congress targeted when it enacted section 513(f)(2) of the FD&C Act as part of the Food and Drug Administration Modernization Act of 1997 (FDAMA). The process created by this provision, which was referred to in FDAMA as the Evaluation of Automatic Class III Designation, will be referred to as the “De Novo classification process”2 throughout this guidance document. Congress included this section to limit unnecessary expenditure of FDA and industry resources that could occur if devices for which general controls or general and special controls provide a reasonable assurance of safety and effectiveness were subject to premarket approval under section 515 of the FD&C Act. Section 513(f)(2) of the FD&C Act has allowed manufacturers to submit a De Novo request to FDA for devices “automatically” classified into Class III by operation of section 513(f)(1). As enacted by FDAMA, in order to submit a De Novo request, a device first had to be found not substantially equivalent (NSE) to legally-marketed predicate devices through a premarket notification (510(k)). The 21st Century Cures Act of 20163 removed a requirement that a De Novo request be submitted within 30 days of receiving an NSE determination.4
Section 513(f)(2) was modified by section 607 of FDASIA, which created an alternative mechanism for submitting a De Novo request that does not require that a device be reviewed first under a 510(k) and found NSE prior to submission of a De Novo request. If a person believes their device is appropriate for classification into Class I or Class II and determines, based on currently available information, there is no legally marketed predicate device, they may submit a De Novo request without a preceding 510(k) and NSE (hereafter “Direct De Novo”).
FDA is issuing this guidance to provide updated recommendations for interactions with FDA related to the De Novo classification process, including what information to submit when seeking a path to market via the De Novo classification process. This guidance replaces “New Section 513(f)(2) – Evaluation of Automatic Class III Designation, Guidance for Industry and CDRH Staff,” dated February 19, 1998.
The De Novo Classification Process
In accordance with section 513(f)(2) of the FD&C Act, you may submit a De Novo request for FDA to make a classification determination for the device according to the criteria at section 513(a)(1) of the FD&C Act. The De Novo request must include a description of the device and detailed information and reasons for any recommended classification (see section 513(f)(2)(A)(v) of the FD&C Act). FDA must make a classification determination for the device that is the subject of the De Novo request by written order within 120 days of the request (see section 513(f)(2)(A)(iii) of the FD&C Act).
If the requester demonstrates that the criteria at section 513(a)(1)(A) or (B) of the FD&C Act are met, we will grant the De Novo request, in which case the specific device and device type is classified in class I or class II. The granting of the De Novo request allows the device to be marketed immediately, creates a classification regulation for devices of this type, and permits the device to serve as a predicate device. We will publish a notice in the Federal Register announcing the classification and the controls necessary to provide reasonable assurance of safety and effectiveness. Note that the classification, including any special controls, is effective on the date the order letter is issued granting the De Novo request. If the De Novo request is declined, the device remains in class III and may not be marketed, unless the device is found substantially equivalent to an existing legally marketed class I, class II, or preamendments device, the device is reclassified under section 513(f)(3) of the FD&C Act, a PMA is approved, or a new De Novo request is granted.
FDA will review De Novo requests for devices that are not within a device type that has been classified under the criteria at section 513(a)(1) of the FD&C Act. This includes devices that do not fall within any existing classification regulation, where the De Novo requester either determines that there is no predicate device or has received an NSE determination on a 510(k) submission. If the device is within a type for which there is an existing classification regulation or one or more approved PMAs, the appropriate mechanism for classification into class I or II would be reclassification under section 513(e) or section 513(f)(3) of the FD&C Act.5
For devices that have already undergone 510(k) review, FDA will consider a De Novo request if the device has been determined to be NSE due to: (1) the lack of an identifiable predicate device, (2) a new intended use, or (3) different technological characteristics that raise different questions of safety and effectiveness. Devices that have been found to be NSE due solely to performance data that is inadequate to demonstrate substantial equivalence (SE) would generally be ineligible for the De Novo classification process.6
In addition, the following criteria should be met for a device for which a De Novo request is submitted:
The device should appear, based on what is known about the device, to meet the statutory standards for classification into class I or class II under section 513(a)(1) of the FD&C Act, i.e., general controls or general and special controls would provide reasonable assurance of the safety and effectiveness of the device; and
You should sufficiently understand and be able to explain all of the probable risks to health and probable benefits of the device, explain the measures needed to effectively mitigate all probable risks, and explain how device safety and effectiveness can be assured through the application of general controls or general and special controls7.
Submitting a De Novo Request for FDA Review
This guidance describes two mechanisms for interacting with FDA regarding a device for which De Novo classification may be appropriate:
Pre-Submission (Pre-Sub). A Pre-Sub is not required in order to obtain FDA review of a De Novo request, but it is a useful way for requesters to obtain early feedback from FDA. A Pre-Sub allows FDA to provide feedback on whether a device may be eligible for the De Novo classification process, including whether a potential predicate device exists, and/or to advise you on the documentation needed in a subsequent De Novo request. The primary advantage of a Pre-Sub is that it provides an opportunity to obtain our preliminary perspective on the likely regulatory controls necessary to provide a reasonable assurance of safety and effectiveness, as well as feedback on the evidence, including non-clinical and/or clinical data, that will likely be necessary to support the De Novo request. By obtaining this feedback, you can optimize your resources in collecting the safety and effectiveness evidence needed to support a De Novo request, without performing additional unnecessary tests. This should also facilitate the review of a subsequent De Novo request.
De Novo Request. A De Novo request may be submitted with or without a preceding 510(k). The success of a De Novo request that is filed without a Pre-Sub will depend more heavily on how well you search for a potential predicate device, identify the risks to health and special controls (if applicable), and provide adequate valid scientific evidence to support granting the De Novo request.
The De Novo review process is outlined in Attachment 1.
In preparing to submit a De Novo request, we suggest you review publicly posted information, including decision summary documents, for recently granted CDRH De Novo requests, available on our website at http://www.fda.gov/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDRH/CDRHTransparency/ucm232269.htm and https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPMN/denovo.cfm or the CBER website (see http://www.fda.gov/BiologicsBloodVaccines/BloodBloodProducts/ApprovedProducts/default.htm)
A
Pre-Sub may be submitted early in the development process for a
device; however, we believe it is most useful after you have
identified the proposed intended use and key aspects of the device
design sufficient to permit a meaningful discussion. A Pre-Sub
related to a future anticipated De Novo request should contain
sufficient information to enable us to provide guidance on the test
methods and protocols that should be used for the collection of
non-clinical and/or clinical data. A Pre-Sub is strongly recommended
prior to the submission of a De Novo request, especially for devices
we have not previously reviewed under a 510(k). De Novo Pre-Subs
will be handled in accordance with our normal pre-submission process.
For information on Pre-Subs, please see the FDA guidance document
entitled “Guidance
for Industry and FDA Staff, Requests for Feedback on Medical Device
Submissions: The Pre-Submission Program and Meetings with Food and
Drug Administration Staff”
(Pre-Sub
Guidance), available at
http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/UCM311176.pdf.
In addition to the recommended content for all Pre-Subs
(device description, proposed intended use/indications for use,
previous submissions, etc.), we suggest that a Pre-Sub prior to
submission of a De Novo request also include:
Proposed Class (I or II). Describe why you believe general or general and special controls are adequate to provide reasonable assurance of safety and effectiveness.
The searches of FDA public databases and other resources, including search terms, used to establish that no legally marketed device and no classification regulation for the same device type exists. Provide a list of regulations, 510(k)s, PMAs, and/or product codes that may be relevant to the subject device. You should also provide a rationale for why the subject device does not fit within and/or is different from any identified classification regulations, 510(k)s, PMAs, and/or product codes, based on available information.
Specific questions regarding review issues relevant to a planned De Novo request. Insofar as they are necessary in order for us to consider the specific questions, the Pre-Sub should also include the following:
Each identified risk to health associated with the device and the reason for each risk (tracing back to risk analysis, clinical testing, etc.). Briefly describe any ongoing and/or planned protocols/studies that need to be completed to collect the necessary data to establish the device’s risk profile.
Information regarding the safety and effectiveness of the device. Cite the types of valid scientific evidence you anticipate providing in your De Novo request, including types of data/studies relating to the device’s safety and effectiveness. Briefly describe any ongoing and/or planned protocols/studies that need to be completed to collect the necessary safety and effectiveness data.
Protocols for non-clinical and clinical studies (if applicable), including how they will address the risks you anticipate and targeted performance levels that will demonstrate that general controls or general and special controls are sufficient to provide reasonable assurance of safety and effectiveness.
The proposed mitigation measure(s)/control(s) for each risk, based on the best available information at the time of the submission. Highlight which mitigations are general controls and which are special controls. Provide details in the Pre-Sub on each recommended mitigation measure (e.g., specific testing required, labeling.).
Examples of questions to pose to FDA in a De Novo Pre-Sub include:
Based on the device description, its intended use/indications for use, and/or technological characteristics, and information on the search performed for legally marketed devices, does FDA believe the device is eligible for De Novo classification?
Are there other risks, in addition to those identified in the Pre-Sub, given the indications for use for the device?
If applicable, are there controls that should be considered to provide a reasonable assurance of safety and effectiveness for the device?
Are the non-clinical study protocols sufficient to allow the collection of data from which conclusions about device safety and/or effectiveness can be drawn? For example:
Is the identified level of concern the appropriate level of concern for the device software?8
What, if any, additional biocompatibility and/or sterility testing would be appropriate?
If clinical data are needed, are the proposed study design and selected control group appropriate?
After you submit your Pre-Sub, we may ask you for clarification or to provide more information. You may also request meetings with us. For more information on Pre-Subs and meetings with FDA staff, please see the Pre-Sub Guidance.
De Novo Request
The De Novo request should include all information and evidence that you are aware of regarding the safety and effectiveness of the device, including the general controls or general and special controls that you believe would provide reasonable assurance of safety and effectiveness. The De Novo request should establish the risk profile of the device, establish the benefits of device use, and provide valid scientific evidence demonstrating the performance characteristics of the device. Attachment 2 contains the suggested content of a De Novo request.
For De Novo requests, sponsors must submit at least one valid electronic copy (eCopy). See section 745A(b) of the FD&C Act and FDA’s eCopy guidance, “eCopy Program for Medical Device Submissions”, available at http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/UCM313794.pdf. De Novo requests (and subsequent submissions, as applicable) submitted without valid eCopies will be placed on hold and the review clock will not start until a valid eCopy is received.
Address for De Novo Requests
For devices regulated by CDRH, De Novo requests should be submitted to:
U.S. Food and Drug Administration
Center for Devices
and Radiological Health
Document Mail Center –
WO66-G609
10903 New Hampshire Ave
Silver Spring, Maryland
20993-0002
For devices regulated by CBER, De Novo requests should be submitted to:
U.S. Food and Drug Administration
Center for Biologics Evaluation and Research
Document Control Center
10903 New Hampshire Ave.
WO71-G112
Silver Spring, Maryland 20993-0002
If, at the end of our review of a 510(k), we determine that a device is NSE due to lack of a predicate, a new intended use or different types of technology issues, we may indicate that the device may be suitable for review under the De Novo classification process. The 510(k) review will occur per standard review practices for 510(k)s and in accordance with current performance goals. If we believe general controls or general and special controls may provide reasonable assurance of safety and effectiveness, we may indicate in the NSE letter that the product may be appropriate for the De Novo classification process under section 513(f)(2) of the FD&C Act. Inclusion of this language within an NSE letter does not indicate that sufficient information currently exists within the 510(k) submission to support a successful De Novo request, but simply indicates that given the risk profile of the device, it seems reasonable that De Novo classification may be appropriate.
Once a De Novo request is received, whether or not it is preceded by a 510(k), we will verify that another submission for the same device (same technological characteristics and same indication(s) for use) from the same requester is not under review (e.g., Pre-Sub, 510(k) or PMA). We will not review two submissions for the same device from the same requester simultaneously. If we identify another submission for the same device, we will place your file on administrative hold. We will not begin review of the De Novo request and will notify you that to start the review, you would need to withdraw the other submission.
We will also check that the content of the De Novo request includes the information required by section 513(f)(2) of the FD&C Act. As provided by section 513(f)(2)(A)(ii) of the FD&C Act, in order to submit a Direct De Novo request, the submitter must determine that there is no legally marketed device upon which to base a determination of substantial equivalence. Under section 513(f)(2)(A)(i) of the FD&C Act, a De Novo request preceded by a 510(k) must be for a device type that has not been previously classified; thus, if you submit a De Novo request after receipt of an NSE determination, you should confirm that no device of the same type has legally entered the market since the time of the NSE determination. See Attachment 2 for discussion of what information you should submit in the classification summary. De Novo requests that lack information to determine whether a potential predicate device exists may be placed on hold. As provided by section 513(f)(2)(v) of the FD&C Act, if you are recommending that your device be regulated as a Class II device, you must also submit an initial draft proposal for applicable special controls.9 If your De Novo request is placed on hold, the review clock stops and we will notify you that it is on hold pending receipt of information regarding potential predicates or a draft proposal for special controls. In the event you do not provide the requested information within 180 calendar days, we will consider your De Novo request to be withdrawn.
Next, we will conduct a classification review of legally marketed device types. We will analyze whether an existing legally marketed device of the same type exists (e.g., whether your device likely falls under an existing Class II classification regulation), including whether a predicate has been recently established through the De Novo classification process. If a likely predicate device exists or your device falls under a class III classification regulation, we intend to decline your De Novo request and notify you of the basis for our decision. If the device falls within a class III classification regulation or there is one or more approved PMAs for the same type of device and we believe general and/or special controls are adequate to provide a reasonable assurance of safety and effectiveness, the appropriate mechanism for classification into class I or II would be reclassification under section 513(e) or 513(f)(3) of the FD&C Act. If no existing legally marketed device of the same type is identified, we will continue our review.
We do not anticipate that De Novo requests for the same device type will frequently be under review concurrently. However, in cases where a De Novo request is granted while another device of the same type is under review in a separate De Novo request, after the first De Novo request is granted, FDA intends to notify the submitter of the other De Novo request still under review that a predicate has been established and that the De Novo request still under review will be declined. The submitter of the declined De Novo request may leverage all information in the De Novo request by incorporating it by reference in a new submission but will still be required to demonstrate substantial equivalence in a subsequent 510(k), including conformity with the newly established special controls for the device type (if Class II).
Upon successful completion of the submission and classification review, FDA will begin the substantive review of the De Novo request. If the De Novo request is missing information and/or data necessary to determine whether general controls or general and special controls can provide reasonable assurance of safety and effectiveness, we may issue an additional information (AI) letter or request information via interactive review. Issuance of an AI letter stops the review clock, and once you provide a complete response, the clock will resume and review will continue. If you fail to provide a complete response within 180 calendar days of the date of the AI request, we will consider the De Novo request to be withdrawn. If a De Novo request is withdrawn due to failure to submit adequate information, a new De Novo request is required in order to reinitiate review of the device under the De Novo classification process.
If general controls or general and special controls are insufficient to provide reasonable assurance of safety and effectiveness or the information and/or the data provided in the De Novo request are insufficient to determine whether general controls or general and special controls can provide a reasonable assurance of safety and effectiveness, we will decline the De Novo request and you may not legally market the device. If a De Novo request is declined, we will issue a written order to the requestor identifying the reasons, including lack of performance data, which warrant declining the De Novo request and the device remains in class III and may not be marketed. You may either submit an application for premarket approval (PMA) under section 515 of the FD&C Act or collect additional information to address the issues and submit a new De Novo request that includes the additional information.
If your data and information demonstrate that general controls or general and special controls are adequate to provide reasonable assurance of safety and effectiveness, we will grant the De Novo request. If a De Novo request is granted, we will issue you a written order granting the De Novo request and identifying the classification of the device (either class I or class II). For class II devices, we will also identify the special controls. Effective on the date of the granting order, the requester may immediately begin marketing the device subject to the general controls and any identified special controls, and the device may be used as a predicate device for future 510(k) submissions as appropriate.
We will then publish a final order in the Federal Register providing public notice of the decision, which will result in codification of the device’s identification, classification, and applicable requirements in Title 21 of the Code of Federal Regulations (device classifications are at parts 862 – 892).
A De Novo request for a device that receives marketing authorization via the De Novo classification process is referred to as having been “granted” (similar to “PMA approved” and “510(k) cleared”).If a De Novo request is granted, we intend to make the written order to the submitter granting the De Novo request and a summary of our review of the De Novo request available on the CDRH website (see http://www.fda.gov/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDRH/CDRHTransparency/ucm232269.htm and https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPMN/denovo.cfm) or the CBER website (see http://www.fda.gov/BiologicsBloodVaccines/BloodBloodProducts/ApprovedProducts/default.htm). Once a De Novo request is granted, then the subject device may be used as a predicate for any future 510(k) submissions. Information posted to the FDA website will be redacted to protect any confidential commercial, trade secret, or personal privacy information in accordance with 21 CFR Part 20.
Paperwork Reduction Act of 1995
This guidance contains information collection provisions that are subject to review by the Office of Management and Budget (OMB) under the Paperwork Reduction Act of 1995 (44 U.S.C. 3501-3520).
The time required to complete this information collection is estimated to average 100 hours per response under 513(f)(2)(i) of the FD&C Act, 180 hours per response under 513(f)(2)(ii) of the FD&C Act, or 10 minutes per response for requests for withdrawal, including the time to review instructions, search existing data sources, gather the data needed, and complete and review the information collection. Send comments regarding this burden estimate or suggestions for reducing this burden to:
FDA
PRA Staff,
Office of Operations,
Food and Drug
Administration,
PRAStaff@fda.hhs.gov
This guidance also refers to currently approved collections of information. The collections of information in the guidance document “Requests for Feedback on Medical Device Submissions: The Pre-Submission Program and Meetings with Food and Drug Administration Staff” have been approved under OMB control number 0910-0756. The collections of information in 21 CFR part 807, subpart E have been approved under OMB control number 0910-0120; the collections of information in 21 CFR part 814 have been approved under OMB control number 0910-0231; and the collections of information in 21 CFR part 801 have been approved under OMB control number 0910-0485.
An agency may not conduct or sponsor, and a person is not required to respond to, a collection of information unless it displays a currently valid OMB control number. The OMB control number for this information collection is 0910-xxxx (expires xx/xx/xxxx).
Attachment 1
De Novo Request Review Process
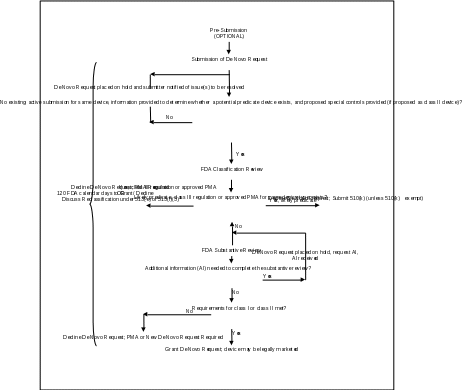
Attachment 2
Recommended Content of a De Novo Request
The cover letter for a De Novo request should clearly identify “De Novo Classification Request”
If significant data for any of
the sections below are contained in a previous submission, you may
provide cross-reference to the information. Any cross-references
should include applicable volume/section/page numbers as
appropriate.
Administrative Information:
Requester name, contact name, address, phone, fax, e-mail.
Regulatory History:
Describe
any prior submissions to FDA for the device, including any 510(k)s
and related NSE decisions, IDEs, Pre-Subs, and/or previously
withdrawn or declined De Novo requests.
For any previous submissions where we provided feedback, please identify how you have responded to the identified issues.
Device Information and Summary:
Provide the device name and a description of all main functions, technological characteristics, components, and accessories. Include a summary of the directions for use/usage instructions.
Indications for Use:
Identify the proposed indications for use (including prescription and/or over-the-counter use). Identify the disease or condition the device will diagnose, treat, prevent, cure, or mitigate, including a description of the patient population for which the device is intended.
Change
Summary (if appropriate):
Describe in detail any changes made to your device or proposed indications since any prior Pre-Sub or 510(k), as appropriate. This summary should include changes to the device, as well as changes to test protocols and/or labeling.
Classification Summary:
For Direct De Novo requests, describe
your search for legally marketed devices of the same type. Provide a
list of classification regulations, cleared 510(k)s, approved PMAs,
and/or product codes that may relate to or are potentially similar to
the subject device. You should also provide a rationale for why the
subject device is different from and/or does not fit within any
identified classification regulations, 510(k)s, PMAs, and/or product
codes, based on available information.
If the same
device (same technological characteristics and same indication(s) for
use) from the same requester has been previously found NSE due to
lack of a predicate, new intended use, or different questions of
safety and effectiveness, the relevant 510(k) number should be
submitted for this section along with a summary of this search
performed since the NSE was issued.
Classification Recommendation (and, for Class II Devices ONLY, Proposed Special Controls):
Provide the recommended class (I or II). Describe why you believe general controls or general and special controls are adequate to provide reasonable assurance of safety and effectiveness. For class II devices, provide proposed special controls along with cross-references to other information within the request demonstrating that the device meets these special controls.
Supporting Protocols and/or Data:
Provide a summary of all non-clinical and clinical testing (if applicable) that provide a reasonable assurance of safety and effectiveness for your specific device and that demonstrate that general controls or general and special controls are sufficient to provide a reasonable assurance of safety and effectiveness. The summary should include the objective of the testing, a description of study design, and a description of the results. For human subject testing, the summary should also describe the study population, selection and exclusion criteria, duration, data collection methodology, observed adverse reactions, and statistical analysis. The summary should include links to appendices, etc., which contain the detailed final protocols and supporting data.
Summary of Benefits:
Provide information supporting the effectiveness of the device. Cite the available data/studies supporting effectiveness. This section may include references to available published literature, where applicable. In providing this information, consider the factors that FDA considers in making benefit-risk determinations. Factors in determining the extent of the probable benefits include the type of benefit, the magnitude of the benefit, the probability of the patient experiencing benefit, and the duration of effect. More information is available in Section 4.1 of the FDA guidance document entitled “Factors to Consider When Making Benefit Risk Determinations in Medical Device Premarket Approval and De Novo Classifications”, available at https://www.fda.gov/downloads/medicaldevices/deviceregulationandguidance/guidancedocuments/ucm506679.pdf.
Summary of Identified Risks to Health:
List each risk to health and identify the reason for each risk (tracing back to risk analysis, clinical testing, etc.). Summarize the studies completed and how they support safety. In considering the extent of the probable risks, FDA considers multiple factors including the severity, types, number, and rates of harmful events associated with the use of the device (including serious adverse events and procedure-related complications); the probability of a harmful event; the duration of harmful events; and, for diagnostic devices, the risk from false-positive or false-negative results. More information is available in Section 4.2 of the FDA guidance document entitled “Factors to Consider When Making Benefit-Risk Determinations in Medical Device Premarket Approval and De Novo Classifications”.
Risk and Mitigation Information:
Provide a table showing the proposed mitigation(s) for each identified risk to health. Identify which mitigations are general controls and which are special controls. Provide specific section and page numbers where the details for each recommended mitigation measure (e.g., specific testing required) can be found in the submission.
Identified Risk |
Recommended Mitigation Measures |
Supporting Data Contained in De Novo |
EXAMPLE: Adverse tissue reaction |
Specified Biocompatibility Testing Requirements (special control) |
Testing in compliance with recognized standard (Section XX, page XXX) |
EXAMPLE: Device failure due to XXX (mechanical failure, software anomaly, use error, etc.) |
Specified Non-clinical Testing (special control), Device Specific Labeling Requirements (special control), Medical Device Reporting (MDR) (general control) |
Test protocols and results (Section XX, pages XXX)
|
EXAMPLE: Failure to properly interpret test results |
Device Specific Labeling Requirements (special control) |
Draft device labeling (Section XX, pages XXX) |
Benefit-Risk Considerations:
Provide a discussion demonstrating that, when subject to general controls or general and special controls, the probable benefits to health from use of the device outweigh any probable injury or illness from such use. More information is available in the FDA guidance document entitled “Factors to Consider When Making Benefit-Risk Determinations in Medical Device Premarket Approval and De Novo Classifications”.
Device Labeling:
Provide device labeling that clearly indicates the proposed intended use and indications for use, limitations, contraindications, etc.10
1 Prior to the Food and Drug Administration Safety and Innovation Act of 2012 (FDASIA), FDA reclassified devices under section 513(e) of the FD&C Act through rulemaking; FDASIA changed this to an order process.
2 The process has been termed “De Novo” because it requires the agency to evaluate novel devices anew, in accordance with the criteria at section 513(a)(1) of the FD&C Act.
3 On December 13, 2016, as part of the 21st Century Cures Act (Pub. L. 114-255), Congress revised § 513(f)(2)(i) of the FD&C Act.
4 For more information regarding demonstration of substantial equivalence, please see the FDA guidance document entitled “The 510(k) Program: Evaluating Substantial Equivalence in Premarket Notifications [510(k)]”, available at https://www.fda.gov/downloads/MedicalDevices/.../UCM284443.pdf.
5 See the Agency’s Reclassification web page at: https://www.fda.gov/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDRH/CDRHTransparency/ucm378724.htm.
6 For more information regarding demonstration of substantial equivalence, please see “The 510(k) Program: Evaluating Substantial Equivalence in Premarket Notifications [510(k)] – Guidance for Industry and Food and Drug Administration Staff” (https://www.fda.gov/downloads/MedicalDevices/.../UCM284443.pdf).
7 For more information on benefit-risk determinations, please see “Guidance for Industry and Food and Drug Administration Staff – Factors to Consider When Making Benefit-Risk Determinations in Medical Device Premarket Approvals and De Novo Classification” (https://www.fda.gov/downloads/medicaldevices/deviceregulationandguidance/guidancedocuments/ucm517504.pdf).
8 For more information on software, please see “Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices” (http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm089543.htm)
9
Per section 513(a)(1)(B) of the FD&C Act, special controls
include “the promulgation of performance standards, postmarket
surveillance, patient registries, development and dissemination of
guidance documents (including guidance on the submission of clinical
data in premarket notification submissions in accordance with
section 510(k) of the act), recommendations, and other appropriate
actions as the Commissioner deems necessary to provide such
assurance.” Typical special controls include specific
performance testing requirements, which may include performance
and/or clinical testing, and labeling requirements.
10 Labeling is defined in section 201(m) of the FD&C Act, 21 U.S.C. 321(m), as “all labels and other written , printed or graphic matter (1) upon any article or any of its containers or wrappers, or (2) accompanying such article.” Labeling may include package inserts, instructions for use (for patient and/or physician, as applicable), service manuals, etc.
| File Type | application/vnd.openxmlformats-officedocument.wordprocessingml.document |
| File Modified | 0000-00-00 |
| File Created | 0000-00-00 |
© 2025 OMB.report | Privacy Policy