Pre-RFD Submissions/Meetings
Draft Guidance for Industry; How to Prepare a Pre-Request for Designation (Pre-RFD)
NEW_Pre-RFD Guidance_7-2017
Pre-RFD Submissions/Meetings
OMB: 0910-0845
Contains Nonbinding Recommendations
Draft — Not for Implementation
How to Prepare a Pre-Request for Designation (Pre-RFD)
____________________________________________________
Guidance for Industry
Document issued on [insert publication date of FR Notice].
The draft of this document was issued on January 13, 2017
Submit one set of either electronic or written comments on this draft guidance by the date provided in the Federal Register notice announcing the availability of the draft guidance. Submit electronic comments to http://www.regulations.gov. Submit written comments to the Division of Dockets Management (HFA-305), Food and Drug Administration, 5630 Fishers Lane, Rm. 1061, Rockville, MD 20852. You should identify all comments with the docket number listed in the notice of availability that publishes in the Federal Register.
Additional copies of this guidance are available from the Office of Combination Products website at http://www.fda.gov/CombinationProducts/default.htm.
For questions on the content of this guidance, contact the Office of Combination Products at combination@fda.gov
-
An agency may not conduct or sponsor, and a person is not required to respond to, a collection of information unless it displays a currently valid OMB control number. The OMB control number for this information collection is [ ] (expires [ ] ).
See additional PRA statement in Section VI of the guidance.
U.S. Department of Health and Human Services
Food and Drug Administration
Office of Combination Products in the Office of the Commissioner
June 2017
TABLE
OF CONTENTS
B. When should I submit a Pre-RFD? 7
C. May I request a meeting with OCP to explain my product? 7
D. How do I submit a Pre-RFD for my product? 8
E. How promptly will FDA review my Pre-RFD? 8
F. May I withdraw my Pre-RFD after submission? 9
G. What if I disagree with OCP’s preliminary assessment? 9
I. Where can I find more information? 9
IV. WHAT INFORMATION SHOULD I INCLUDE IN A PRE-RFD? 9
A. What is the basic information that I should include in my Pre-RFD? 9
C. What format should I follow for my Pre-RFD? 11
D. Does FDA accept electronic submissions for Pre-RFDs? 11
E. How should my Pre-RFD be addressed if I submit it via standard mail? 11
F. Is there a page limit for Pre-RFDs? 12
VI. PAPERWORK REDUCTION ACT OF 1995……………………………………….. 12
VII. APPENDIX 14
This
guidance is intended to assist sponsors in obtaining a preliminary
assessment from the U.S. Food and Drug Administration (FDA or Agency)
through the Pre-Request for Designation (Pre-RFD) process.
Specifically, this guidance explains the Pre-RFD process at the
Office of Combination Products (OCP) and helps a sponsor understand
the type of information to provide in a Pre-RFD. The Pre-RFD process
is available to provide informal, non-binding feedback regarding the
regulatory identity or classification of a human medical product as a
drug, device, biological product, or combination product. In
addition, this informal process provides information about a
non-combination or combination product’s assignment to the
appropriate Agency Center (Center for Drug Evaluation and Research
(CDER), Center for Devices and Radiological Health (CDRH), or Center
for Biologics Evaluation and Research (CBER)) for premarket review
and regulation.
Additional information on topics outside the scope of this guidance may be found on our website (at http://www.fda.gov/CombinationProducts/default.htm.) These topics include the definitions of a non-combination and combination product, as well as information about the formal Request for Designation (RFD) process.
Since its establishment on December 24, 2002, OCP has served as a resource for sponsors at various stages of development of their product. Sponsors often seek OCP feedback on whether their medical product will be regulated as a drug, a device, a biologic, or a combination product, and which FDA medical product Agency Center (CDER, CBER, or CDRH) will regulate it, if it is a non-combination product, or will have the primary jurisdiction for the premarket review and regulation of the product, if it is a combination product.
There
are two ways that a sponsor can receive such a feedback from OCP.
One option is to submit an RFD to receive a formal, binding
determination for the sponsor’s product with
respect to classification and/or center assignment that may be
changed under conditions specified in Section 563 of the FD&C Act
and 21 CFR 3.9 in the regulations. The RFD process is codified in 21
CFR Part 3, and OCP has issued a guidance about this process (see
“How to Write a Request for Designation” at
http://www.fda.gov/RegulatoryInformation/Guidances/ucm126053.htm).
A second more flexible option is for a sponsor to submit an inquiry
to OCP to receive a preliminary jurisdictional assessment, which is
not binding.
Many sponsors seek to utilize the flexibility of more approachable ways to interact with OCP and the medical product Agency Centers to obtain feedback from the Agency before submitting a marketing application to the Agency. Over time, these informal methods of obtaining feedback have become increasingly customary with sponsors, and for some, even preferable to the formal RFD process. Accordingly, FDA is enhancing the transparency and consistency of this process, which will now be called the “Pre-Request for Designation (Pre-RFD) Program.”
This
guidance describes this structured process with clear recommendations
for sponsors wishing to submit Pre-RFDs. It also provides the
process for review of Pre-RFDs by FDA staff, the general timeframes
for sponsors to receive feedback from OCP, and the process for
scheduling teleconferences and meetings in relation to a Pre-RFD.
FDA’s guidance documents, including this guidance, do not establish legally enforceable responsibilities. Instead, guidances describe the Agency’s current thinking on a topic and should be viewed only as recommendations, unless specific regulatory or statutory requirements are cited. The use of the word “should” in Agency guidance documents means that something is suggested or recommended, but not required.
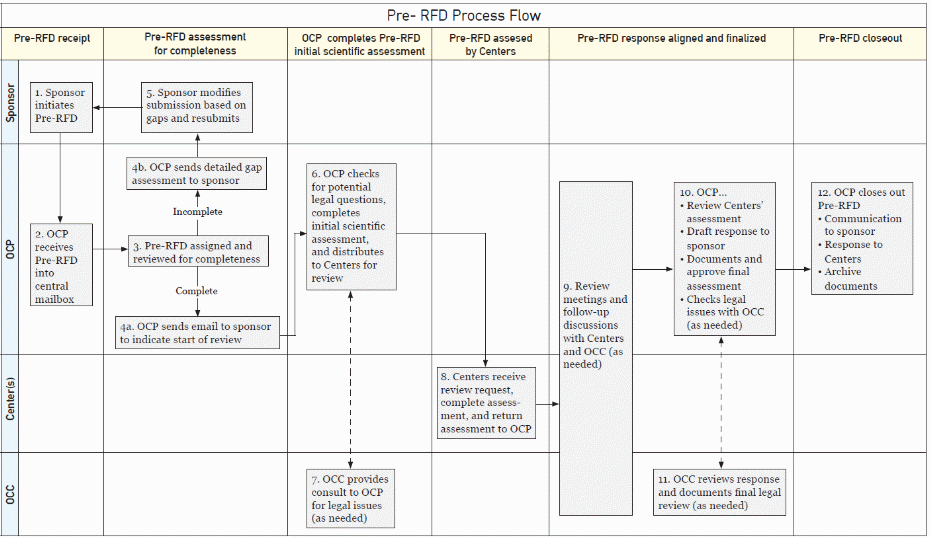
GENERAL INFORMATION REGARDING THE PRE-RFD PROCESS FOR NON-COMBINATION AND COMBINATION PRODUCTS1
A Pre-RFD is a clear and concise written submission that a sponsor may submit to OCP to request FDA’s preliminary, nonbinding assessment of (1) the regulatory identity or classification of a product as a drug, device, biological product, or combination product, and/or (2) whether CBER, CDER, or CDRH will regulate the product if it is a non-combination product, or which of those Agency Centers will have primary jurisdiction for the premarket review and regulation, if it is a combination product.
OCP will provide a written preliminary classification and/or jurisdictional assessment of the product based on the information provided in a specific Pre-RFD. Our goal is to provide you with our feedback within 60 calendar days of receipt of the basic information that you should provide, and we will communicate with you during our review as needed. You can also contact us at any time during the review to ask questions or get clarification. Moreover, if it appears that we will not be able to complete our review within 60 calendar days after receipt of the information, we will communicate the need for more review time to you in a timely manner. Please note that if you change your product significantly after submitting the Pre-RFD to us, such as changing its indication or ingredients, OCP’s feedback may no longer be applicable. Accordingly, we recommend that you contact OCP if you make such a change, since you may need to submit a new Pre-RFD for OCP’s consideration. Additionally, if you have classification or assignment questions regarding multiple related products or product families that have different configurations, ingredients, and/or proposed uses or indications, we recommend submitting a separate Pre-RFD for each product.
Though our Pre-RFD feedback is not binding on the Agency or sponsor, it is OCP’s intent to provide the best advice possible based on the information provided in the Pre-RFD. OCP’s review and written feedback will be made after an in-depth review, and it will contain a thorough and detailed rationale for our assessment. Moreover, our review will include involvement from the relevant Agency Centers, as well as the Office of Chief Counsel when necessary.
The following table highlights some of the key similarities and differences between the RFD and Pre-RFD processes.2 Regarding similarities, in both processes certain basic information is needed for FDA to provide an assessment of the classification and assignment of a product. The basic information includes, for example, a description of a product, proposed use or indications, and a description of how a product achieves its intended therapeutic/diagnostic effects. In addition, some information required for an RFD may optionally be provided as part of a Pre-RFD. For example, a sponsor may wish to provide relevant data/studies, a description of related products, and/or a sponsor recommendation. Differences between the RFD and Pre-RFD process include length of submission and depth of regulatory analysis that a sponsor must provide. For example, RFDs cannot exceed 15 pages, but no such length requirement exists for Pre-RFD submissions. Moreover, for an RFD, a sponsor must provide an analysis of a product’s classification, a primary mode of action (PMOA) analysis (if the RFD concerns a combination product), and a recommendation regarding jurisdictional assignment. Such information is optional in the Pre-RFD process (additional details are provided in Section IV.A. and in Appendix A). We note that FDA would conduct the same type of PMOA analysis in a Pre-RFD as in a RFD regardless of whether a sponsor chooses to make a recommendation regarding the PMOA of its combination product in a Pre-RFD.
.
Submission Type |
RFD (Information Required) |
Pre-RFD (Information Recommended) |
Description of Product |
Yes |
Yes |
Proposed Use or Indications for Use |
Yes |
Yes |
Description of the manufacturing processes, including the sources of all components |
Yes |
Optional (if available; recommended if it is an HCT/P or a biological product) |
Supportive Data/Studies |
Yes |
Optional (if available) |
Description of how a product achieves its intended therapeutic/diagnostic effects |
Yes |
Yes |
Analysis of Classification, Primary Mode of Action (PMOA), if it is a combination product, and Jurisdictional Assignment |
Yes |
Optional (if available) |
Description of Related Products |
Yes |
Optional (if available) |
Sponsor Recommendation |
Yes |
Optional (if available) |
Page limit |
Yes |
No3 |
A Pre-RFD may be submitted at any point during medical product development.
A Pre-RFD is especially beneficial when the classification of a product or the Agency Center to which it should be assigned is unclear or in dispute and your product is very early in its development, or if you are contemplating whether or not to pursue a specific configuration or a specific indication. Understanding how FDA regulates your product and the appropriate regulatory pathway to market for your product will help lead to better decision-making for your company.
We note that a Pre-RFD is not necessary for every product. For example, some products have clearly established classification and jurisdictional assignments. OCP has published a list of various examples of these types of products on its website at combination@fda.gov.
We encourage you to contact OCP before submitting your Pre-RFD if you have any questions, including if you are uncertain about the type of information to include.
In most cases, the Agency will be able to make its preliminary assessment based on a well-written Pre-RFD. However, if you believe a meeting with OCP would be helpful in providing the Agency with a better understanding of how the product works, you can request a meeting either prior to or after submission of a Pre-RFD. If you request a meeting after submission of a Pre-RFD but prior to our feedback, please keep in mind that such a meeting will potentially extend the timeframe of review of your product, so that we can adequately consider issues raised during the meeting.
If you request a meeting, you should include an explanation of the issues you would like to address and any supportive information. Please note that meetings involving subject matter experts from two or more Agency Centers may take longer to schedule due to the increased number of participants, the availability of conference rooms, and the added complexity that exists for many combination products. When possible and appropriate, OCP encourages you to consider a teleconference instead of an in-person meeting.
Generally speaking, FDA will need about four weeks to review information submitted as part of the meeting request prior to the meeting. This timeframe will give OCP and the necessary subject matter experts from the Agency Centers adequate time to review, comment, and possibly follow-up on any issues prior to your meeting. Therefore, it is very important that you provide complete background information at the time of your initial meeting request. If you wish to supplement your background information package with any new or modified information after this date, we may have to reschedule the meeting or delay our feedback on certain discussion topics related to the new or updated information. While the importance of a complete background package cannot be overstated, it should also be noted that submission of extraneous information can be counterproductive. In order for us to provide the most efficient and beneficial advice to you, please keep your background information targeted and focused on the issues in your Pre-RFD that you would like us to consider.
Meeting requests may be submitted by writing to OCP at the address listed at the front of this guidance and below in Section IV.E. or by e-mail to combination@fda.gov. Please label your correspondence as “Pre-RFD Meeting Request.”
Part III of this document addresses the recommended format and specific content of a Pre-RFD in detail. You may submit a Pre-RFD, and include any relevant data pertaining to your product, which should be clearly labeled as such, to combination@fda.gov or to the address listed below in Section IV.E.
Within 5 business days of its receipt of a Pre-RFD, OCP aims to review the Pre-RFD submission to ensure that it has adequate information for us to make our preliminary assessment. OCP will then either send you an acknowledgement email that our assessment will proceed, or we will detail the additional information needed before we can begin our assessment.
OCP’s goal is to provide feedback on a Pre-RFD within 60 calendar days after we receive complete information to begin our review. However, please keep in mind that the speed of the review depends on the quality and adequacy of the information submitted. Furthermore, if you choose to submit a large amount of data for FDA to consider, it may be necessary for us to take longer than our goal of 60 calendar days to fully consider the information provided.
Yes,
you may withdraw your Pre-RFD by notifying OCP in writing, via email
or correspondence sent to the address below in section IV.E., any
time after its submission and before FDA issues its preliminary
assessment.
If
you disagree with OCP’s preliminary assessment of your product,
you may contact our office to discuss our findings. After receiving
our feedback, should you wish to present additional information or
new data that was not presented in the original Pre-RFD, we encourage
you to contact our office to discuss such additional information. We
will be happy to provide you with assistance before you submit a new
Pre-RFD. Once you submit a new Pre-RFD to us, OCP will consider
this Pre-RFD a new submission and provide it with a new review and
thorough assessment. Alternatively, you can submit an RFD for our
consideration (see 21 CFR Part 3 and the guidance document “How
to Write an RFD”
for additional information on the necessary information, format, and
page limit for an RFD). Once your RFD has been reviewed for
completeness and accepted for filing, we will review the RFD and
provide,within 60 calendar days of filing, a binding determination
with respect to classification and/or center assignment that may be
changed under conditions specified in Section 563 of the FD&C Act
and 21 CFR 3.9 in the regulations. Under 21 CFR 3.8(c), you may
request reconsideration of an RFD decision within 15 calendar days of
receipt of the designation letter. FDA will review and act in
writing within 15 calendar days of our receipt of the request for
reconsideration. Alternatively,
once you receive your RFD decision, you may directly appeal that
decision to the Associate Commissioner of Special Medical Programs
pursuant to 21 CFR 10.75.
Contact
information for OCP, including mailing address, e-mail address, phone
number, and fax number are listed below in Part IV.E.
More information about product classification and assignment, as well as the regulation of combination products, is available on the OCP website at https://www.fda.gov/CombinationProducts/default.htm .
Contact information including your name, company’s name, email address, and telephone number.
A complete description of the product and, if applicable, the following information.
The 510(k), PMA, NDA, ANDA, BLA, or any other FDA regulatory submission number associated with the product.
Name of the product and all component products; and
A photo/diagram of the product
For products sourced from biologically-derived materials, describe how the material was processed and a characterization of the identity of the final product.
An explanation of how the product works. And, though optional, you may include additional information describing details (i.e., study conditions/methods, identification of controls, results and conclusions) of relevant testing that supports how the product works. Please be aware that comparisons to other products or biocompatibility testing are typically not helpful in understanding how the product works.
An explanation of how the product will be marketed. For instance, will the product have separately marketed constituent parts that are to be labeled for use together, or will it have components that either will be physically or chemically combined to make a single entity or will be co-packaged?
A listing of all components/ingredients, including the amount and reasoning for including each component/ingredient, in the product. If the product contains a solution/liquid/gel/powder, please provide a listing of all ingredients (active and inactive), their amount/concentration, and the reason for including the ingredient in the product.
Proposed use/intended use/indications for use statement.
Instructions for use/conditions of use.
All known methods of action and the mechanism(s) by which each is achieved.
For products that might be combination products, any information that you might have, if any, to support the relative contribution of different components to the overall intended therapeutic/diagnostic effects of the combination product. Though optional, you may provide a detailed description of any supporting tests/studies if such information is available and you would like FDA to consider the information.
A list of claims that you intend to make or have made regarding the product.
What if I have additional information about my product that I would like to share with FDA in my Pre-RFD?4
In addition to the above requested information needed for FDA to conduct a Pre-RFD review, you may also choose to provide additional information about a product. Information beyond that which is listed in Section IV.A is optional. However, in the event that you have prepared other information that you wish us to consider, we would be happy to do so. This information might include, for example, a recommendation for lead Agency Center, if your product is a combination product, or any products that you believe may be similar to your product, or any other information that you consider relevant for us to consider during our preliminary assessment.
We
recommend that you provide the information concisely set forth in the
screening checklist in the Appendix and explained in detail in the
above sections. It would be helpful if you identify each section of
the Pre-RFD in a separate heading that correspond to those listed in
the screening checklist followed by your response. We also recommend
you use a standard typeface (e.g., Times New Roman), which should be
in an easily readable font size (e.g., 12).
Yes. You may submit your Pre-RFD by e-mail to combination@fda.gov in a common electronic format, such as a Portable Document Format (PDF) or a Word Document. As explained in the preceding section, only one electronic submission is necessary.
We encourage you to submit your Pre-RFD electronically to our inbox at combination@fda.gov. However, if you choose to submit your Pre-RFD via standard mail, you should send it to the following address:
Office
of Combination Products
Office of the Commissioner
Food and
Drug Administration
WO32 Hub/Mail Room #5129
10903 New
Hampshire Avenue
Silver Spring, MD 20993-0002
(Phone): 301-796-8930
(Fax): 301-847-8619
Combination@fda.gov
In order for the Pre-RFD to be sent to the correct location, the envelope should be clearly marked as a “Pre-Request for Designation.” Only one Pre-RFD submission is necessary. In other words, no copies of your original Pre-RFD submission are required.
No, there is no page limit for Pre-RFDs (nor is there a word limit for emailed Pre-RFD submissions that are not in PDF or Word format). However, we encourage you to limit your Pre-RFD to a succinct summary of information relevant for OCP to make its preliminary assessment.
This guidance provides our recommendations for the format and content of a Pre-RFD submission. We recommend you pay particular attention to these sections of your Pre-RFD as applicable:
A complete description of the product, including its composition (what is your product?);
The intended use/indications for use of the product (why would your product be used?);
The methods of action of the product (how does your product work?);
If your product may be a combination product, information you may have, if any, regarding the relative contribution of each of its components to the overall intended therapeutic/diagnostic effects of the combination product; and
Any claims you have made or plan to make about your product.
OCP is always available as a resource to you. We strongly encourage you to contact OCP before submitting your Pre-RFD if you have any questions, or if you are uncertain about the type of information to include.
This guidance contains information collection provisions that are subject to review by the Office of Management and Budget (OMB) under the Paperwork Reduction Act of 1995 (44 U.S.C. 3501-3520).
The time required to complete this information collection is estimated to average 13 hours per response, including the time to review instructions, search existing data sources, gather the data needed, and complete and review the information collection. Send comments regarding this burden estimate to:
Department of Health and Human Services
Food and Drug Administration
Office of Chief Information Officer
Paperwork Reduction Act (PRA) Staff
PRAStaff@fda.hhs.gov
An agency may not conduct or sponsor, and a person is not required to respond to, a collection of information unless it displays a currently valid OMB control number. The OMB control number for this information collection is [ ] (expires [ ]).
VII. Appendix:
:
Contact information including your name, company’s name, email address, and telephone number.
A complete description of the product and, if applicable, the following information.
The 510(k), PMA, NDA, ANDA, BLA, or any other FDA regulatory submission number associated with the product.
Name of the product and all component products; and
A photo/diagram of the product
For products sourced from biologically-derived materials, describe how the material was processed and a characterization of the identity of the final product.
An explanation of how the product works. Though optional, you may provide additional information describing details (i.e., study conditions/methods, identification of controls, results, and conclusions) of relevant testing that supports how the product works. Please be aware that comparisons to other products or biocompatibility testing are typically not helpful in understanding how the product works.
An explanation of how the product will be marketed (e.g., kit configuration). For instance, will the product have separately marketed constituent parts that are to be labeled for use together, or will it have components that will be physically or chemically combined to make a single entity or co-package?
A listing of all components/ingredients, including the amount and purpose for including each component/ingredient, in the product. If the product contains a solution/liquid/gel/powder, please provide a listing of all ingredients (active and inactive), their amount/concentration, and the reason for including each ingredient in the product.
Proposed use/intended use/indications for use statement.
Instructions for use/conditions of use.
All known methods of action and the mechanism(s) by which each is achieved.
For products that might be combination products, information that you might have, if any, to support the relative contribution of different components to the overall intended therapeutic/diagnostic effects of the combination product. Though optional, you may provide detailed description of any relevant tests/studies if such information is available and you would like FDA to consider the information.
A list of claims that you have made or plan to make regarding the product.
1 For the purposes of this guidance document, parties who submit a Pre-RFD to the Agency are referred to as “sponsors,” “you” or “your”; the terms “we,” “us,” and “our” refer to FDA staff from the Office of Combination Products.
2 Please note that the information listed in this Pre-RFD Guidance does not include all the necessary information required for an RFD. If you wish to submit an RFD, you should consult 21 CFR 3.7 as well as the guidance about what to include in an RFD (see “How to Write a Request for Designation” at http://www.fda.gov/RegulatoryInformation/Guidances/ucm126053.htm).
3 Although there is no page limit for Pre-RFDs, we encourage you to limit your Pre-RFD to a succinct summary of information relevant for OCP to make its preliminary assessment.
4 We note that to the extent any information a sponsor provides in a Pre-RFD is trade secret or confidential commercial information, such information is protected by the same confidentiality laws as those that protect such information provided in an RFD.
| File Type | application/msword |
| Author | Leigh Hayes |
| Last Modified By | SYSTEM |
| File Modified | 2017-07-25 |
| File Created | 2017-07-25 |
© 2026 OMB.report | Privacy Policy