HPHC2 RIHSC Protocol
HPHC2 RIHSC Protocol 12.07.2016.docx
Generic Clearance for the Collection of Qualitative Data on Tobacco Products and Communications
HPHC2 RIHSC Protocol
OMB: 0910-0796
D EPARTMENT
OF HEALTH AND HUMAN SERVICES
EPARTMENT
OF HEALTH AND HUMAN SERVICES
_________________________________________________________________________________________________________
Food and Drug Administration
Center for Tobacco Products
10903 New Hampshire Ave
Silver Spring, MD 20903
Protocol Title: Consumer Comprehension of Displays of Harmful and Potentially Harmful Constituents (HPHCs) in Tobacco Products
Protocol Version or Date: 11-30-2016
Study Mechanism, Research Team, and Credentials of Investigators
Identify the mechanism for conducting the study (e.g., contract, IAA, MOU, etc.), its ID number, and attach any associated instrument: Contract: HHSF223201110005B, Order: HHSF22316001 (See attached contract).
Contractor Info:
RTI International
3040 Cornwallis Road
Research Triangle Park, NC 27709
Telephone: (301) 770-8219 (x2-8219)
Point of Contact:
Jennifer Alexander, JAlexander@rti.org
Office of Research Protection, Institutional Review Board
Federalwide Assurance No. 3331
Ina Wallace, PhD, Chair
FDA Sponsor : Katherine Margolis, PhD (Role: FDA Research Lead)
Additional FDA researchers:
Jennifer Bernat, PhD (Role: FDA Research team)
Matia Johnson (Role: Project Manager)
David Portnoy, PhD MPH (Role: FDA Research team)
Principal Investigator: Jonathan Blitstein, PhD (RTI)
Additional RTI Study Staff:
Jennifer Alexander (Role: Interviewer, data analysis)
Peyton Williams (Role: Interviewer)
Bonnie Hepler (Role: Notetaker, data analysis)
Wendi Elkins (Role: Notetaker)
Abstract
CTP is seeking to conduct a series of 50 individual in-depth interviews (IDIs) with adults and youth (14 to 17 years old) to gather information about different ways of presenting harmful and potentially harmful constituent (HPHC) information. The purpose of the research is to gain insight on consumer comprehension of and preferences regarding presentation of information about HPHCs in tobacco products and tobacco smoke which will inform the Agency’s efforts to implement the mandatory publicly available list of HPHCs required by the Family Smoking Prevention and Tobacco Control Act (the Tobacco Control Act). Interview participants will be asked to answer questions regarding different ways of presenting HPHC information by brand and by quantity in each brand and subbrand.
The study will consist of a series of IDIs with adults and youth. IDIs will be designed to answer the following questions:
• What do adults and youth know and what do they perceive about HPHCs in tobacco products?
• How and in what formats do adults and youth prefer to receive information regarding HPHCs?
• How do adults and youth understand and perceive the information about HPHCs provided in sample formats?
Regulatory and Scientific Significance of the Research
Describe the regulatory background and significance of the research.
On June 22, 2009, President Obama signed the Family Smoking Prevention and Tobacco Control Act (Tobacco Control Act) into law. The Tobacco Control Act amended the Federal Food, Drug, and Cosmetic (FD&C) Act by adding a new chapter granting the US Food and Drug Administration (FDA) important new authority to regulate the manufacture, marketing, and distribution of tobacco products to protect the public health generally and to reduce tobacco use by minors. Section 904(e) of the FD&C Act requires FDA to establish, and periodically revise as appropriate, ‘‘a list of harmful and potentially harmful constituents (HPHCs), including smoke constituents, to health in each tobacco product by brand and by quantity in each brand and subbrand.’’ Section 904(d)(1) of the Act further requires that the list be published in a format that is understandable and not misleading to lay persons.
To fulfil this mandate, FDA’s Center for Tobacco Products (CTP) requires a qualitative study that builds on prior research to gain insight on consumer comprehension of information about HPHCs in tobacco products and tobacco smoke, which are chemicals or chemical compounds in a tobacco product or tobacco smoke that cause, or could cause, harm. Examples of HPHCs include toxicants, carcinogens, and addictive chemicals and chemical compounds.
CTP will conduct a qualitative study to gather information about different ways of presenting HPHC information by brand and by quantity in each brand and subbrand in a format that is understandable and not misleading to a layperson.
Identify the CTP research priorities addressed
The research in the proposed study will inform regulatory activities defined by the Tobacco Control Act. Specifically the research addresses the following priority: Effective communication strategies regarding harmful and potentially harmful constituents and risks of tobacco products.
Objectives and Hypotheses
The goal of this study is to gather information about different ways of presenting HPHC information. Results will be used to inform strategies to effectively communicate about HPHC information that is understandable and not misleading to the layperson.
Study Design and Methods
Inclusion criteria for subjects, samples or data, as applicable
Adults = Age 18+, Youth = 14-17 years
Adult Cigarette User
Adult Smokeless User
Adult Former Tobacco User
Youth Cigarette and/or Smokeless User
Youth Susceptible to Tobacco Use
Exclusion criteria for subjects, samples, or data, as applicable
Below 14 years of age
Does not match tobacco use or susceptibility criteria
Youth without parental permission to participate
Has a medical or non-medical condition that hinders his/her ability to read and/or understand written materials in English
Has in the past 5 years worked for a tobacco or cigarette company; a public health or community organization involved in communicating the dangers of smoking or the benefits of quitting; a marketing, advertising, or public relations agency or department; the Federal Government including the U.S. Food and Drug Administration (FDA), the National Institutes of Health (NIH), the Centers for Disease Control and Prevention (CDC), the Substance Abuse and Mental Health Services Administration (SAMHSA), or the Centers for Medicare & Medicaid Services (CMS) or someone in household has in the past 5 years worked for any of these
Has ever lobbied on behalf of the tobacco industry, or someone in household has lobbied on behalf of the tobacco industry
Has ever represented or worked on behalf of a tobacco company in connection with a tobacco lawsuit, or someone in household has done so
Has participated in any paid market research in the past 6 months
The number of subjects to be studied, per arm and total, if applicable, along with the rationale for the sample size(s).
A total of 65 participants to complete a total of 50 IDIs will be recruited. This overrecruitment by 15 participants is designed to ensure that the required 50 are completed.
Study design, including the controls, blinding, or randomization to be used, if any, and the methods used to minimize bias on the part of subjects, investigators, and analysis
Formative Qualitative Research Design: The present study consists of IDIs and is, therefore, qualitative in nature and does not incorporate controls, blinding, or randomization.
Describe all study procedures in the order in which they will be performed or affect subjects
Adult participants will be identified by staff at subcontracted recruitment facilities using the facilities’ existing proprietary databases of those who have previously agreed to be contacted. Using the screener, recruiting staff will contact and will ask eligible participants if they’d like to be a part of the study, and identify the date and time of the IDI (see attached screener).
Youth participants will be identified by recruiting staff using their database of youth whose parents have previously agreed to be contacted. Recruiting staff will contact the parent, present the study, then ask to speak to the youth. Using the screener, recruiting staff will ask eligible youth participants if they’d like to be a part of the study, and identify the date and time of the IDI (see attached screener). Youth will be required to bring with them to their IDI a signed parental permission form (reading level grade 8) (see attached parent permission form). Individual youth responses to screening question will not be shared with parents.
Recruitment Plan and Screening Procedures
Recruitment is led by a different subcontracted recruitment facility in each location. Adults and youth (ages 14-17), male and female, from all ethnic/racial backgrounds are eligible to participate. The recruitment facility draws from their existing proprietary database of individuals interested in research participation. The IDIs are tentatively scheduled to be conducted in Pensacola, FL, Birmingham, AL, and San Diego, CA. The recruitment facilities have demonstrated capability in recruiting individuals from diverse demographic backgrounds. The recruitment screener includes questions related to educational attainment, race/ethnicity, and tobacco use behaviors (see Screener). These questions are used to ensure that there is an appropriate and diverse racial/ethnic representation, a range of ages, and a mix of educational attainment. Because the present study employs IDIs, it is qualitative in nature and does not incorporate controls, blinding, or randomization. Participants will be recruited via telephone screening procedure. If the individual is eligible for participation, the individual will receive logistical and contact information for the study. Youth participants will also receive a parent permission form that must be signed and presented before participating in their IDI.
Upon arrival at the facility, participants will be greeted, parent permission forms will be collected from youth, and all participants will be given a consent (adult) or assent (youth) form to read. The interviewer will answer any questions about the consent/assent form, and participants will sign if they wish to continue (see attached consent and assent forms). Signing the consent/assent form indicates agreement with being audio recorded and video streamed.
Participants will then begin the interview. All interviews will be conducted with a trained interviewer in the room with the participant. Participants will be able to skip any questions they do not want to answer. The measures were created to address specific research questions, and are open-ended.
IDIs will not last more 60 minutes. The interviews will be led by a trained interviewer who will use a semi-structured interview guide to lead the discussion (see Interview Guide). IDIs will be audio-recorded and will be web-streamed over a secure site (Focus Vision) for the sponsor to attend remotely (see more detail below under Section XVII) if needed and available. Video streams will not be recorded. Participants will be informed that they will be audio recorded and video streamed in the consent/assent forms. By signing the consent/assent, they consent/assent to audio recording and video streaming.
At the end of the discussion, participants will be debriefed and compensated for their time by the facility staff ($40 for adults, $20 for youth and an additional $20 for parents/guardians who bring youth based on OMB guidelines in cash, check, or prepaid bank card depending on facility). Participants who attend the interview but choose not to participate at any point in the process will be compensated at the same level.
Describe the tobacco cessation message and optional cessation intervention or referral you will employ for tobacco users, including its content, format, and timing. Alternatively, justify not including such a message.
At the conclusion of the session, participants will be told “I would like to thank you for coming here today and participating in this discussion. This research was sponsored by the Food and Drug Administration also known as the FDA. FDA would like to thank you for sharing your opinions as they will be very useful in helping them to understand people’s reactions and thoughts about the tobacco products we have talked about. The FDA wants you to know that there is no safe tobacco product, including the products we talked about today. Here is a pamphlet with information from FDA on how users can quit.”
Product Information
Participants will not use any tobacco products during the course of this study.
Duration of the Study
Data collection should take approximately three months. Each interview will take no more than 60 minutes. Each participant will only be involved in a single interview and no follow-up will occur. Recruitment will start within two weeks after OMB and IRB approvals are received. Interviews will be conducted over the course of three months. The study will be complete upon receiving a summary of methodology and findings from RTI.
Location
The research is tentatively scheduled to take place in market research facilities or hotels in Pensacola, FL, Birmingham, AL, and San Diego, CA.
Analysis of the Study
Data Analysis & Reporting
In general, interview research relies on qualitative methods and is not intended to yield results that are statistically projectable. Analysis of IDI data will be conducted using a thematic analysis of transcripts and notes. Transcripts will only contain participant first names. The methodology and findings report provided by the study contractor shall include the study purpose, methods, description of the participants, major findings from the IDIs, and any differences detected in responses between the various segments. The report will contain no information by which participants can be identified.
Subject Selection and Recruitment
This study is not meant to produce generalizable findings. Participants will be recruited by a market research company. They will use their database and a screening questionnaire to identify participants who may be eligible for the study.
Provide the rationale for research subject selection based on the gender /ethnicity/race/socioeconomic categories at risk for the condition under study.
All genders, ethnicities, races, and socioeconomic categories can participate in the interviews if they meet the inclusion criteria.
Provide the rationale for subgroupings of research subjects, if applicable
As these will be individual interviews, participants will not be subgrouped.
Risk to Benefit Ratio
Describe any potential risks, whether physical, psychological, social, economic, legal, or other as a result of participation in your research study.
There are minimal psychological, social, or legal risks to participating in this study. Participants will be asked to share their attitudes and opinions; however, the topic is not sensitive in nature. Participation is voluntary, and participants can choose not to answer any of the questions.
Assess the likelihood and seriousness of the potential risks.
Any serious risks are extremely unlikely. Participants may be uncomfortable discussing things like their tobacco use behavior but can choose to leave the interview or not answer any questions.
Describe procedures you will utilize to minimize any risks, and their likely effectiveness
No personally identifiable information (PII) will be collected by RTI or FDA or stored with interview data, so no PII will be tied to any participant. Participants can leave an interview or refuse to answer any question at any time.
Describe potential benefits
There are no direct benefits from participating in this study. Participants’ opinions will help improve FDA’s understanding of how people think about and use tobacco products.
Discuss the circumstances and procedures for terminating subject participation, under both voluntary and involuntary circumstances, including who may make the decision and who communicates with the subject about this issue.
Participants may leave the IDI at any time. They need to only state their desire to leave once the interview initiates. Participants will receive compensation regardless of the length of time of their participation. If the participant seems to be intoxicated or under duress, the interviewer may choose to conclude the interview early based on discussions with FDA observers. The participant will be kindly excused and fully compensated.
Make the case (summative statement) for study approval based on the justified risks to benefits within the context of the knowledge to be gained.
There is little to no risk involved with this study.
Screening Tests and Interview Prior to Subject Enrollment
See attached screener.
Compensation, Incentives and Rewards for Participation
Describe the type(s), amount(s), and schedule of incentive(s) or reward(s).
One time payment at the market research facility of $40 for adults, $20 for youth, and $20 for parents/guardians who transport youth.
The amount of the incentive for participants is $40 for adults, $20 for youth, and $20 for parents/guardians who transport youth.Incentives in cash, check, or prepaid bank card (depending on facility procedures) will be provided at the conclusion of the IDI. Participants have the right to terminate their participation at any point, without penalty. If an individual must leave or is asked to leave for any reason before the conclusion of the session, he/she will receive the full incentive amount. Individuals who arrive late for their scheduled time may not be able to participate. These individuals will not be given a consent form, are not considered participants, and thus will not receive an incentive.
Informed consent (IC)
The informed consent process will happen when participants arrive to the facility. The interviewer will give a paper adult consent form (reading level grade 8) or youth assent form (reading level grade 8) and pen, and will be available to answer any questions. Written consent or assent will be documented and stored securely by RTI in a locked file cabinet for 3 years.
Data and Safety Monitoring [Adverse Events, Unanticipated Problems, Protocol Deviations, Violations, and Study Monitoring] Detail your plan for monitoring individual research subjects for protocol compliance and adverse health effects
There are no anticipated adverse events, serious adverse events, or unanticipated problems. Participants may end involvement at any time. CTP-RHISC will be notified of any protocol violations or subject complaints. Investigators will take action to respond as appropriate. Market research facilities will inform the sponsor immediately of any adverse events or complaints. In the event of a confidentiality breach, which is unlikely because PII and PPI will never be stored with interview data, the sponsor will report any confidentiality breaches within one hour of discovery:
all reviewing / overseeing IRBs: FDA RIHSC: RIHSC@fda.hhs.gov; RTI’s Office of Research Protection orpe@rti.org.
FDA Sponsor: Katherine Margolis, Katherine.Margolis@fda.hhs.gov
CTP RIHSC
The sponsor will attend as many interviews as possible in-person. If the sponsor cannot make the interviews in person, the sponsor will participate via live web-streaming if available.
Data Management, Subject Privacy, and Data Confidentiality (Ref: 45 CFR §46.111(a)(7))
Discuss your data collection and management procedures
The subcontracted recruitment facilities will have participants’ names and contact information but will not share this contact information with RTI or FDA. Study staff (RTI and FDA) will only receive first names and last initials of participants on all materials. Consent forms will be signed by participants. All consent forms will be retained by RTI in a locked file cabinet for 3 years.
Audio recordings will be used to create transcripts of all interviews. RTI subcontracts with a transcriptionist. Only first names of participants will be transcribed and any PII inadvertently disclosed will be redacted from all transcripts. Transcripts are then shared with FDA. Once all of the interviews are completed and transcriptions are completed, audio recordings are destroyed.
Screening details that include PII are not retained, nor will such information ever be given to RTI or FDA. Last names of the participants are never used on any study materials (typed lists of participants, name plates, transcripts, draft or final reports). The data are always used in the aggregate. Consent will be obtained from adult participants. Assent will be obtained from youth participants. In order to be eligible to participate, youth must bring with them to the interview a signed parental permission form. Interviews will be audio recorded only. Once all of the interviews are completed and transcriptions are completed, audio recordings are destroyed. Interviews will also be live web-streamed over a secure server but will not be recorded.
After a review of the proposed study protocol and data management procedures, RTI can confirm that there is no confidential or personally identifiable information in an electronic format. The only electronic records that RTI will receive include de-identified transcripts of participant interviews. The inadvertent mention of PII by participants during the interview will be redacted from transcripts. Accordingly, there appears to be no risk of an electronic data breach.
RTI will maintain signed informed consent document on premises in locked files for three years as required. In the event of a data breach, RTI will follow appropriate procedures that include timely notification of any potentially affected participants, notification of RTI’s Privacy Officer, and complete documentation of the breach and subsequent actions.
The following is documentation of RTI’s confidentiality procedures:
In this section, RTI’s approach to maintaining the confidentiality of study data is described. These data include information provided directly by respondents as well as information obtained about respondents from other sources. RTI has the following legal foundation for confidentiality and confidentiality procedures:
i. Confidentiality
The following measures are standard protocol used to ensure confidentiality: (1) Last names of the participants are never used on any materials (e.g. typed lists of participants, reports); (2) Reports do not contain any personally identifying information and are stored securely on a password-protected computer; (3) Quotes that might be used in the memorandum to illustrate findings are not attributed to the individual. No names are used in any reporting, when direct quotations are used, participants are identified by their segmentation (for example: youth, smoker, San Diego).
Signed consent forms are stored by RTI for 3 years in a locked filing cabinet. Audio recordings are destroyed after being transcribed. Transcripts will be sent electronically to FDA and stored on RTI password protected servers at RTI for 3 years.
ii. RTI’s Confidentiality Statement
Employing appropriate confidentiality procedures and presenting them to potential study respondents are essential to the success of RTI research studies, especially those seeking personal or identifiable data. Our confidentiality procedures are designed to implement safeguards to assure potential study respondents that their data are protected should they decide to participate in the research study or should their information be used for research purposes.
As part of the informed consent process for every RTI study, potential study participants are informed about
• the purpose of the data collection,
• the types of information to be obtained,
• the persons or entities who will have access to their information, and
• the fact that all information provided by the respondent will be kept confidential to the fullest extent of the law.
iii. Legal Foundation for Confidentiality
Government at all levels has acted to ensure research subjects’ rights to privacy and confidentiality, and these rights are protected by numerous laws and regulations. RTI is committed to complying with the statutes and regulations relating to data collection that safeguard the confidentiality of responses, as demonstrated by the following:
• RTI researchers conform to all requirements of the Privacy Act of 1974.
• Under sections 45 CFR 46 and 21 CFR 50 and 56 of the Code of Federal Regulations, the RTI Institutional Review Boards (IRBs) evaluate research projects to ensure that human subjects are protected and that confidentiality procedures are adequate.
• Under 42 USC Section 241(d), RTI will seek authorization to protect the privacy of study subjects by withholding their names and other identifying characteristics from all unauthorized persons.
RTI staff is familiar with these federal provisions. Some staff also have experience complying with the laws and regulations of individual states and agencies governing the subjects’ rights to privacy and confidentiality.
In addition to the legal requirements listed above, RTI will take other steps to secure the confidentiality of study data. In most studies, RTI promises respondents that their data will be treated as confidential and released to the public only in the form of aggregate statistics that cannot be associated with any individual or household. RTI does not have the legal precedent for confidentiality that doctors, lawyers, and the clergy have. In certain studies, there will be exceptions to confidentiality that include information reported by respondents about child abuse or neglect, imminent harm, communicable diseases, or suspected terrorist activities (under the Patriot Act). This information will be reported to the proper authorities, according to applicable state laws. Respondents are told in the informed consent that this information will not be kept confidential.
iv. RTI’s Confidentiality Procedures
RTI is dedicated to maintaining the confidentiality of all information on human subjects, particularly sensitive or identifiable data. Because disclosure of research data could have serious consequences for research participants, RTI manages information about human subjects in ways that prevent unauthorized access to study data at any time during the study. Our confidentiality guidelines specifically limit the release of personally identifying information about participants. Generally, these identifiers are kept in separate data files and are not released to anyone outside the project team. Respondents must be fully informed about how and to whom their information is released. Each project staff member is required to uphold our confidentiality guidelines and procedures. The primary responsibility for protecting the confidentiality of subjects rests with the project director, who oversees the proper implementation of the procedures and who institutes any changes necessary to ensure confidentiality.
The confidentiality procedures that we routinely implement at RTI to maintain the confidentiality of study data are listed below.
• The principal investigator must describe for the IRB the confidentiality and data security measures to be used in the study as part of the IRB approval process.
• The confidentiality protections must be described in the informed consent form that participants sign.
• The principal investigator provides the necessary instruction on confidentiality requirements and procedures to all project staff.
• Each staff member involved in any phase of handling sensitive personal information is required to sign a legally binding confidentiality agreement. (Both existing staff and newly hired personnel working on the contract must sign the agreement.)
• Field staff are required to sign a pledge of confidentiality that reinforces confidentiality requirements of the study and states that any procedural violation that jeopardizes a respondent’s privacy will be grounds for immediate termination and possible legal action.
• Staff members (both field and on-site) are informed that they must not discuss any aspects of an individual study case with anyone who is not directly involved in the project, and that discussions among colleagues should take place only when necessary for the accurate and timely completion of work.
• Access to hard copy or electronic data is restricted to authorized staff members.
• Electronic data are stored in a location within the RTI network that provides the appropriate level of security based on the sensitivity or identifiability of the data.
• Staff members are not allowed to interview or process data for subjects they know personally.
• The disclosure of personal identifiers to outside sources requires the subject’s specific consent (unless disclosure is required by state or federal law)
Screening forms used by the recruiters will not be given to the contract staff at RTI. No screening data will be retained by the market research facilities, nor will such data ever be turned over to FDA. After IDIs are completed, screening forms will be shredded. Consent forms will be collected by interviewers/ RTI staff and stored at RTI International in a locked file cabinet. Interviews will be audio-recorded and stored on the RTI International encrypted network. Summary documents as well as de-identified transcripts will be sent to FDA.
REFERENCE LIST
APPENDIX/ATTACHMENTS
See attached documents:
Screener
Parent permission form
Consent form
Assent form
Interview guide
Sample formats
APPENDIX
Sample Formats

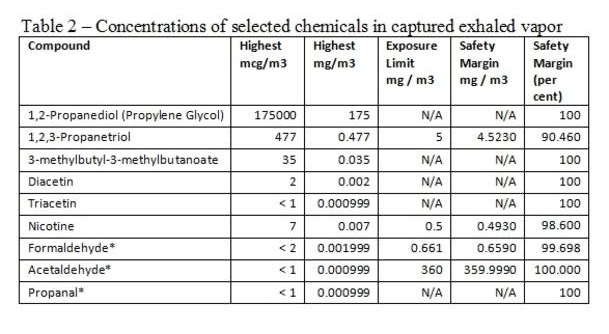

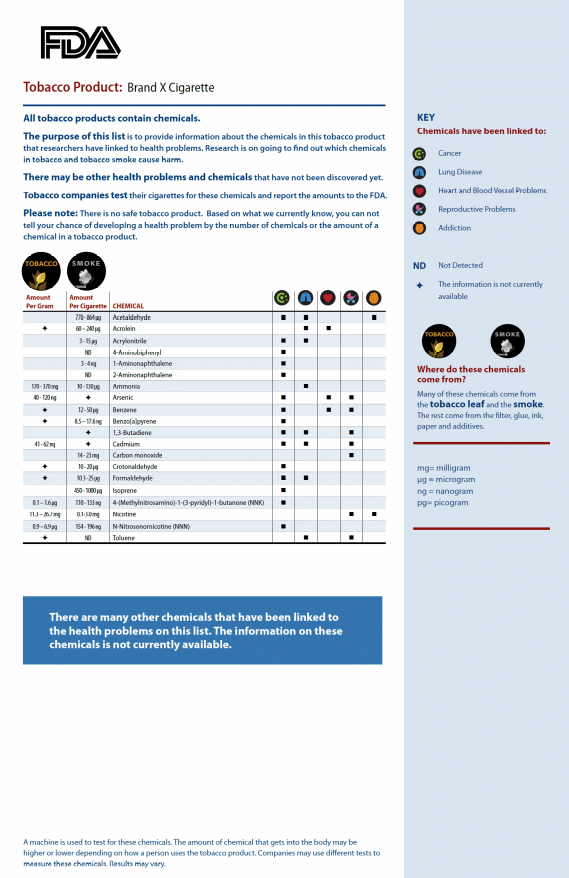
| File Type | application/vnd.openxmlformats-officedocument.wordprocessingml.document |
| File Title | DHHS Letterhead |
| Subject | Letterhead - To Customize |
| Author | JBowers |
| File Modified | 0000-00-00 |
| File Created | 2021-01-26 |
© 2026 OMB.report | Privacy Policy