8-29-07 memo
OMB Review Responses 070815_final.doc
Evaluation of a Medication Therapy Management Program to Improve Patient Safety in Medicare Beneficiaries
8-29-07 memo
OMB: 0935-0136
1. A2:
a. what are the “discrepancies” that will be found through the “medication reconciliation process”? If the patient is self-reporting all medication use, what kinds of discrepancies do you expect to find?
We apologize for the confusion over the term “discrepancies,” as defined below in question 1b. It appears that throughout the protocol, the term “discrepancies” was used in two distinct ways: 1) as a way of describing a problem between the intent of the prescriber and the way a patient is taking their medication, as identified by the MTM clinician; and 2) as a formal outcome of the study with a formal definition.
From the supporting statement, p3, last paragraph:
For this study, medication reconciliation is defined as a process of building a complete medication list based on the most current information taken from prescription bottle directions, patient interview, and medical record information with the goal of reducing medication errors. Discrepancies will be identified as part of a formal process of DRP assessment. Discrepancies and other DRPs will be brought to the attention of the prescriber, and if appropriate, changes made to the existing medication regimen. As a minimum, medication reconciliation will be accomplished by building a list based on the patient’s medication bottles. Otherwise, a complete medication reconciliation process entails assessing the available medical record, gathering the most recent medication changes as documented by physician(s), comparing information to medication bottles and updating the patient’s medication list.
To clarify, the term “discrepancies” used in the context above would be differences between the medications (and directions) the provider intended the patient to take and what the patient actually took, as identified by the MTM clinician. In identifying these discrepancies, the MTM clinician does not have access to the patient chart (other than the provided clinical synopsis for the enhanced MTM group) or other sources of information that may identify the prescriber’s intent. Still, the MTM clinician may identify occasions where there appears to be discord between the way a patient is taking their medication and the usual recommended use of the medication or directions appearing on the clinical synopsis provided to the MTM clinician (enhanced MTM group only).
The second manner in which “discrepancies” is used is as a formal outcome of the study (p13, Hypothesis 2.2). In this manner, the definition of “discrepancies” is strictly adhered to. Prescriber’s intent is identified through a “Best Possible Medication History (BPMH).” Patient medication use will be determined through the MTM clinician’s records of how a patient is actually taking their medications from the patient interview. Differences between these two lists will be labeled as “discrepancies.”
b. please explain the differences between “discrepancies,” “adverse medication events,” “drug related problems.” Sometimes they seem to be used as distinct concepts, other times they seem to be used as if they are all synonymous.
Discrepancy: differences from the way a patient should be taking their medications (usually derived from a thorough review process that identifies the prescriber’s intent) and how a patient is taking their medications (usually derived from a patient interview process).
Drug Related Problem: an event or circumstance involving drug therapy that actually or potentially interferes with desired health outcomes.
According to the tool that we are using to identify DRPs (the PCNE classification for drug related problems), the basic classification of DRP has 6 primary domains for problems, 6 primary domains for causes and 5 primary domains for interventions. Problems are adverse reactions, drug choice problems, dosing problems, drug use problems, interactions, and “other” problems.
Adverse Drug Event: harm caused by a drug or the inappropriate use of a drug.
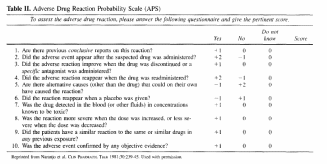
Functionally, we will be assessing ADEs by telephone interview, using a two step process. The first step is to identify “symptoms” using a tool by Jarernsiripornkul et al. The second step is to determine if the symptom is potentially drug related. This will be accomplished using the Naranjo algorithm (also called the Adverse Drug Reactions Probability Scale or APS). The algorithm is shown below and is described under question 5b:
c. will study subjects receive instructions on what they are expected to do (e.g. the patient diary) as well as what they are expected to look out for (i.e. will “adverse medication event” be defined for them?)
At the first study visit, patients will be given a visit log to assist them in tracking emergency room, hospital, and doctor’s office visits. They will be given instructions on how to use the log. They will also be informed that, if they want, they can indicate that they believe their problem was due to one of their medications (adverse event). This will assist patients in completing the Patient Visit Telephone Questionnaire (Appendix Q) and the Telephone Interview Questions for Assessing Adverse Drug Events (Appendix G). We will be using the telephone questionnaire by Jarernsiripornkul et al, coupled with the Naranjo algorithm to assess ADEs and do not expect patients to make their own determination of whether their symptom was an ADE.
d. on page 5 of the supporting statement under “Arm 3,” it says that it will take only 10 minutes to obtain a “clinical synopsis” on the basis of reviewing sections of the subjects’ medical records. However, on page 3, it says that a complete medication reconciliation would be unduly burdensome because it would require assessing the patient’s medical records. Please explain this apparent inconsistency. And if a clinical synopsis can be done in only 10 minutes, isn’t it worth doing for Arm 2 as well?
The clinical synopsis (MTM Clinical Documentation Tool; Appendix B) is a 10-minute review and transcription of the medical records onto a paper form to assist the MTM clinician in providing the MTM intervention. We are instructing our research assistants to transfer the clinical synopsis information from the chart to our documentation tool in 10 minutes to simulate the process as it might occur in a community physician’s office.
The supporting statement on page 3 states “As a minimum, medication reconciliation will be accomplished by building a list based on the patient’s medication bottles. Otherwise, a complete medication reconciliation process entails assessing the available medical record, gathering the most recent medication changes as documented by physician(s), comparing information to medication bottles and updating the patient’s medication list.” The reason for limiting information to the MTM clinician was not based on burden, but was purposely imposed in the study to improve the study’s external validity. The study has been designed to mimic how an MTM program would be administered in a community pharmacy. Community pharmacists do not typically have access to patient medical records, and therefore must work with limited information. Our two study intervention arms are designed to assess the benefits of having more information (clinical synopsis vs. no clinical synopsis) on creating better medication lists. These lists will be compared with a Best Possible Medication History (which will include assessing the available medical record, gathering the most recent medication changes as documented by physician, and information from medication bottles) to determine how many discrepancies occur between each intervention method and the BPMH. This is one of our primary outcomes.
The clinical synopsis is the only difference between Arms 2 (basic MTM) and 3 (enhanced MTM). This type of chart information is not typically available to community pharmacists, as they do not have access to a patient’s electronic medical records. As one of our primary outcomes, we are studying whether having this additional information actually enhances the decision making ability of the MTM clinician. If this information is useful (i.e., successfully improves decision making) and efficient, then it might be reasonable for community pharmacists to request this information from physicians’ offices (via fax or other communication method) prior to meeting with their MTM patients.
e. what questions will be used to screen subjects for eligibility for this study? For example, I can’t find any question that asks subjects whether they already participate with their own Part D MTM program.
Patient eligibility criteria are as follows:
1) Be at least 65 years old at enrollment
2) Primarily uses English language for written and oral communication
3) Have three or more comorbid conditions associated with increased healthcare utilization (see Table 1).
4) Have visited a physician at one or more of the clinics on a regular basis (defined as two or more clinic visits over one year prior to the study start) for these conditions.
5) Have received 8 or more different chronic prescription medications over the six months prior to the enrollment period.
6) Have a telephone line and agree to maintain it for at least six months.
7) Have one of the following situations placing him/her at risk for a DRP:
a) Any ER visit in past 30 days or Urgent Care visit in past 30 days leading to a change in medication or change in medication dose
b) New physician visit in past 30 days
c) Hospitalization in past 30 days
d) Invasive procedure (a procedure requiring substantive changes to medication taking practices or which requires informed consent) in past 30 days
e) Change in medication in past 30 days
f) Three or more providers seen in the past year
Patients with the following exclusion criteria will not be enrolled in the study:
1) Terminal condition, where life expectancy is less than 6 months
2) Patients already enrolled in an MTM program where medication reconciliation and/or assessment of DRPs has occurred in the previous 12 months
Note that patients who are already enrolled in a Part D MTM program will be eligible to participate in this study, provided they meet all of the other criteria for inclusion and do not have any exclusion criteria. Patients enrolled in Part D MTM programs do not necessarily have a medication reconciliation conducted as part of their MTM program.
There are three times when patient eligibility information is collected:
1) Prior to enrollment, and after an appropriate HIPAA waiver for chart review has been secured, scheduling and billing records for each participating institutions will be screened for patients age > 65, availability of address and phone number (for contacting patients regarding enrollment), presence of 2 or more clinic visits in the past year, and ICD-9 billing diagnoses for 3 or more chronic conditions from table 1 above.
2) Study investigators will randomize the list of names and contact patients (first by mail and then by phone within 10 days; see Appendixes I and J), to determine patient eligibility for and interest in participating in the study. The telephone screening script includes the following questions:
1. What is your date of birth?
2. Do you have any of the following medical problems?
a. Diabetes YES NO
b. Heart failure YES NO
c. Asthma YES NO
d. High blood pressure YES NO
e. High/abnormal Cholesterol YES NO
f. Emphysema (COPD) YES NO
g. History of heart attack, heart blockage (e.g., stent placement or bypass surgery) YES NO
h. poor kidney function YES NO
i. arthritis YES NO
j. depression YES NO
k. memory problems YES NO
l. chronic pain YES NO
m. take blood thinner (warfarin/Coumadin) on a daily basis YES NO
3. Can you tell me the medications, including over the counter vitamins or supplements, that you take every day? [If patient needs assistance, ask if permission to access their electronic health record and review the last clinic note].
4. Has your doctor changed your medication dose or added a new medication within the past month? YES NO
IF YES: Date of medication change
5. Have you seen a new doctor in the past month? YES NO
IF YES: Date of new provider visit
6. Have you been seen in the Emergency Room or Urgent Care clinic in the past month? YES NO
IF YES: Date of ER/Urgent care visit
7. Have you been discharged from the hospital in the past month?
Note: This questionnaire may be revised to include 2 additional questions about whether the patient has a terminal condition (life expectancy less than 6 months) and whether they have had a medication reconciliation (explained in lay language as a time when they have been asked to bring all of their medications to a meeting with a pharmacist, nurse, or physician and provided a list of the medications they should be taking) done in the previous 12 months.
3) At the baseline study visit, just prior to obtaining informed consent, the research assistant will verify that the patient meets the inclusion and exclusion criteria by running through a checklist. This information was not explicitly stated in the protocol, but will be done as part of the enrollment process. The baseline study visit information will be recorded and used to determine true patient eligibility for the study. The other times that screening information is collected (billing records and telephone screen) are intended to eliminate non-eligible patients before they are asked to travel to the study site for the enrollment visit.
f. who are the people who will be “MTM clinicians” in this study? What are their qualifications? How will they be selected/recruited for this study? It would seem that they would have to be fairly sophisticated if they are going to be expected to assess the extent to which patients are UNTREATED for indications (i.e. they would have to be able to diagnose conditions).
The Medicare Prescription Drug, Improvement and Modernization Act stated:
“A medication therapy management program described in this paragraph is a program of drug therapy management that may be furnished by a pharmacist and that is designed to assure, with respect to targeted beneficiaries described in clause (ii), that covered part D drugs under the prescription drug plan are appropriately used to optimize therapeutic outcomes through improved medication use, and to reduce the risk of adverse events, including adverse drug interactions… Such program shall be developed in cooperation with licensed and practicing pharmacists and physicians.”
This rule has been interpreted to mean that an MTM may be furnished by a pharmacist but may also be furnished by another provider (physician, nurse, etc) within their appropriate and legal scope of practice. We have been cautious throughout the document to ensure that we are not endorsing one professional over another, and thus have used the term MTM clinician. Essentially, it is the intervention that we are testing, and not the abilities of the MTM clinician.
The clinicians that have been hired to conduct this intervention are pharmacists. They will be practicing within their scope of practice and within the requirements of their state laws. All have completed pharmacy practice residencies. Additional training will be provided to the pharmacists, consisting of:
1) An orientation to the Medicare Modernization Act, especially as it relates to Medication Therapy Management, in non-technical language. This training will include the intended purpose of the law, proposed goals of Medication Therapy Management, and a brief overview of the study’s goals, as a type of Medication Therapy Management program.
2) An orientation to the patient care process outlined in the study and the forms that will be used to document patient interactions, patient record keeping, and MTM clinician – primary care provider communication and documentation.
3) A brief overview and review of managing patients who are older and have multiple chronic conditions, including providing a list of medications that generally should be avoided in elderly patients.
The pharmacist clinicians in this study will be very capable of identifying untreated indications – an activity well within the scope of practice of these individuals. In order to assess drug therapy for untreated but known conditions, it is not necessary that the MTM clinician be able to diagnose (e.g., a patient has been told by her physician that she has high blood pressure, but stopped taking the medication because it made her feel dizzy; this is now an untreated condition). Untreated conditions could be identified from the patient interview or from the clinical synopsis. Unfortunately, untreated conditions are a common Drug Related Problem, and are included as one of the domains in the PCNE classification: “Drug Choice Problem: Patient gets or is going to get a wrong (or no drug) drug for his/her disease and/or condition.”
2. A10: has this study received all necessary IRB approvals?
Duke and Baylor/RTI have received the necessary IRB approvals. UIC has submitted for IRB approval and has responded to questions and comments from the IRB. It is anticipated that approval at UIC will be obtained within the next few weeks.
3. A10: all uses of the word “confidential” should be changed to “confidential to the extent permitted by the Privacy Act.”
All uses of the word confidential have been changed as requested.
4. A12: why aren’t you collecting the burden estimates associated with extracting data from patients’ records, analyzing data from records, and telephone interviews? This would seem to be useful information for the public to know if they were envisioning implementing a similar type of MTM (i.e. it will give them an idea of the administrative burden in terms of cost and time to implement this kind of an MTM).
As noted above, we have instructed our research assistants to take no more than 10 minutes to extract data from patient records. We therefore felt that collecting this information was unnecessary. However, as noted above, such information may be of interest to those implementing similar programs and we can collect this information if we are required.
The time burden on the pharmacists of reviewing the clinical synopses is being collected using the MTM Clinician Time Log (Appendix N).
Analysis of the data and collection of information from the telephone interviews are strictly for study purposes and would not be part of a publicly available MTM program. Therefore, collection of time burden estimates for these items would not likely be helpful to the public and would increase the documentation burden of the investigators. Again, we can collect this information if we are required.
5. A16:
a. how will you consider random effects in your regression models?
Generalized Linear Mixed Models (GLMM) were proposed to test the hypotheses in the evaluation of a MTM program. GLMM combine the ideas of generalized linear models with the random effects modeling ideas. In particular, the analysis will be conducted using logistic and Poisson regression models in our study. The purpose of using the random effects is to assess how much variability within each variable of the study sites, clinicians and patient characteristics contributes to the total variation of the dependent variables.
Random effects such as study sites, clinicians and patient characteristics will be added with fixed effects (eg. demographic variables). Each random effect is assumed to have a random Gaussian distribution with zero mean and a constant variance in the proposed models. We thus have four variance components: there are three variances for random effects and a variance of measurement error in each model. We will measure the proportions of the total variability of the dependent variable that is accounted for by the variability of the random effects. We will then determine whether or not the final model includes the random effects.
There are various ways of fitting GLMM in R and SAS. For example, R has MASS and lme4 packages, which use the Penalized Quasi-Likelihood method and lmer function, respectively.
b. what is the “Naranjo algorithm”?
The Naranjo algorithm or APS is a short questionnaire (10 questions) that systematically analyzes various criteria that help establish a causal association between a drug and an adverse event. The 10 questions deal with the following factors: (1) pattern of response, (2) temporal sequence, (3) dechallenge, (4) rechallenge, (5) altemative
causes, (6) placebo response, (7) drug levels in body fluids or tissues, (8) dose-response relationship, (9) experiences of previous patients with the drug, and (10) confirmation by objective evidence (e.g., laboratory tests or direct clinical observation versus patient
report). Each question can be answered as positive, negative, or unknown/not applicable and is scored from - 1 to +2 accordingly. The sum of the numbers ranges from -4 to 13 and indicates the strength of the causal relationship as follows: definite, >=9; probable,
5 to 8; possible, 1 to 4; and doubtful, <0 association.
c. is 6 months enough time to assess the frequency of drug related problems? Presumably, unless a patient is prescribed a new medication during the study period, the patient has been taking these medications for many years (i.e. they are medications for chronic conditions). As such, wouldn’t the physician have already caught any big adverse side effects?
Note that drug related problems are those problems identified by the pharmacist at the time of the medication reconciliation. Six months should be sufficient time to assess adverse drug events. We are enrolling patients who have either had a recent change to their medications or have had a transition in their setting (e.g., a recent ED visit or hospitalization). Adverse drug events are most likely to occur soon after such a change or transition. Evidence to support this comes from a recent study which found that within one month of being discharged from the hospital, 11% of patients had preventable ADEs and 8% were readmitted to the ED or hospital.(Schnipper et al. Arch Int Med 2006; 166:565-571) Furthermore, the patients in our study will be at an increased risk for adverse events because of other identified risk factors that serve as our inclusion criteria, such as high number of medications, multiple chronic conditions, multiple providers, and advanced age. Our method of detecting ADEs has been validated and was designed to be more sensitive than previously used ADE detection methods. For all of the above stated reasons, we expect a 6 month follow-up to be sufficient to assess the frequency of ADEs.
d. it might be useful/interesting to include the Beers list of age-inappropriate drugs as one of the contraindications you look for.
In the Supporting Statement A p.6, we describe the MTM clinician training program. Item 3 states: “A brief overview and review of managing patients who are older and have multiple chronic conditions, including providing a list of medications that generally should be avoided in elderly patients.” This list of medications is the Beers’ list.
Note that the Beers’ list is not a list of contraindicated medications in the elderly. Rather, it is a list of potentially inappropriate medications, initially developed in nursing home patients and recently expanded through an expert panel process. As such, we will be expecting our MTM clinicians (and also the patients’ primary care physicians) to treat these medications as “potentially inappropriate” and to make a risk/benefit assessment when any of these medications are being prescribed. They will not be automatic contraindications and their use will not be automatically considered a DRP. Furthermore, for any given patient, the MTM clinicians may identify drugs other than those on the Beers’ list that may be potentially inappropriate.
e. How will you determine what is a valid “potential drug related problem?”
We are not attempting to validate the MTM clinician and primary care physician’s clinical judgment of what is and is not a DRP. The ability of pharmacists to identify DRPs compared with an expert panel has already been demonstrated.(Isetts et al. Arch Intern Med 2003; 163: 1813-1820.) In this study, expert panelists agreed with pharmacist assessments in 94.2% of the cases, were neutral in 3.6%, and disagreed in 2.2% of cases. We will be comparing the number of DRPs identified by the pharmacists with and without additional chart information in the form of a clinical synopsis. This information is typically not available to community pharmacists, but may improve their ability to identify true DRPs.
f. Will you report the actual and potential drug related problems separately or as one total? Suggest keeping them separate.
DRPs are defined as events or circumstances involving drug therapy that actually or potentially interferes with desired health outcomes. Since we are not measuring patient health outcomes, and are intervening before the desired outcome is affected, it is not possible to identify which DRPs are actual and which are potential. As such, DRPs will not be separated into actual and potential DRPs.
g. When collecting patient medical records data, does this assume that the patient only sees providers at the study sites? Or will the researchers collect medical records from off-site providers as well?
We are not collecting medical records from off-site providers. All 3 study sites are comprehensive academic medical centers with very few out-of-system office visits. Out-of-system emergency health care visits would most likely be referenced in the electronic medical records by the patient’s PCP.
For the study outcomes of ED visits and hospitalizations, we are relying solely on subject report of these occurrences. We expect recall bias to be low, as these are major events, subjects are being provided with logs, and we are assessing these outcomes twice in the 6-month period (at 3 and 6 months).
h. Patient satisfaction questions: how are the questions for 3.1 and 3.2 different? The questions used for 3.1 do not seem specific to identifying “satisfaction with medication regimen,” but rather seem to assess satisfaction will overall quality of care. If the 3 questions for 3.2 have been validated, perhaps this is all you need to ask?
Hypothesis 3.1 is misstated. It should read: “Patients enrolled in an MTM program will have a higher level of satisfaction regarding the care received from their pharmacist compared to the control group, and there will be an additional increase in satisfaction among patients receiving the enhanced MTM intervention.” This modification has been made to the revised supporting statement.
The questions in the PCQ (Hypothesis 3.1) are different from those in Hypothesis 3.2, which focus on the overall satisfaction with healthcare received. Most importantly, the items in the PCQ are specific to the satisfaction of care received by their pharmacist (MTM clinician). The overall satisfaction questions will help determine if changes in the satisfaction with the pharmacist translate to overall improvements in satisfaction with healthcare received.
i. Where is it explained to the subjects that the midpoint of the PCQ scale is equivalent to no change compared with the community pharmacy where the patient normally obtains his/her drugs?
The instructions for describing this part of the survey were omitted the supporting statement. They have been added.
6. B3: besides refusal data, what other analysis will you conduct to assess non-response bias?
Since this is a prospective, randomized, controlled study (RCT) . As such we do not expect “non-response” to affect our ability to determine the impact of our intervention in this study. As with other RCTs, we expect a high degree of internal validity. That is, the results can be trusted given the group of patients that were included. Non-response bias typically refers to surveys where there is concern that responders may differ systematically from non-responders. What the reviewer may be referring to is how refusal to participate in this study may somehow affect our study’s external validity (or generalizability). This of course is a concern for any clinical trial. Other than refusal data, we will not be attempting to determine if those who choose not to participate in this study are substantially different from those who do. However, we will be able to characterize the patients that did participate and our results should only be considered valid for that patient population.
7. surveys: please revise the race/ethnicity questions to comply with OMB standards
The race/ethnicity questions have been revised.
8. response to comments: the response indicates that AHRQ will not consider medication adherence in this study. Isn’t it an important confounder? Also, please respond to comments from Medco (faxed to Doris on July 31).
Poor adherence is considered a drug related problem (as part of the PCNE tool we are using to assess DRPs), and is being identified by the MTM clinicians in the study. However, adherence is not an explicit outcome for a number of reasons. Poor adherence is a patient behavior. Modification of that behavior typically requires lengthy, complex, multifaceted interventions that are beyond the scope of this study as it has been designed.(Kripalani S, Yao X, Haynes RB. Interventions to enhance medication adherence in chronic medical conditions: a systematic review. Arch Intern Med. 2007 Mar 26;167(6):540-50.) Furthermore, programs focusing on altering chronic medication taking behavior have had mixed results. Finally, assessment of adherence is complex. Since we do not consistently have access to patient prescription drug claims, assessment of adherence would likely have to occur via patient self-report which is an unreliable method of assessing adherence.
Since we are randomizing patients in this study, we expect the level of non-adherence to be equally distributed between the three study groups. Therefore, adherence should not be a significant confounder in our study.
9. consent form:
a. if the baseline study visit will mostly happen right after providing informed consent—and if subjects need to have all their medications with them at the baseline study—does that mean that all subjects will have to have all their medication with them at the time of providing consent?
Yes. Subjects will be advised at the end of the “Patient Telephone Screening and Invitation to Participate Script” that they are to bring all of their medications with them to the initial study visit. If patients agree to participate in the study, the first MTM clinic visit time will be scheduled. For convenience, we will allow study subjects randomized to one of the intervention arms to schedule their first MTM clinic visit immediately after the first study visit.
b. Please explain this sentence: “after information about you is given to anyone outside the study, it may be re-disclosed and may no longer be protected by federal privacy laws.”
The consent form that was submitted as part of the OMB Supporting Statement was a form that combined both the Informed Consent and the Authorization to Use and Disclose Health Information (Authorization), being piloted by the UIC IRB. The documents have now been separated into two separate forms, as requested by the UIC IRB. The newly revised consent form does not contain the above language. This language is required as part of (and is still included in) the Authorization form by the U.S. Department of Health and Human Services a part of the Standards for Privacy of Individually Identifiable Health Information under 45 CFR Parts 160 and 164.
c. Please explain this sentence: “this permission to access your medical records has no expiration date…”
The consent form that was submitted as part of the OMB Supporting Statement was a form that combined both the Informed Consent and the Authorization to Use and Disclose Health Information (Authorization), being piloted by the UIC IRB. The documents have now been separated into two separate forms, as requested by the UIC IRB. The newly revised consent form does not contain the above language. This language is required as part of (and is still included in) the Authorization form by the U.S. Department of Health and Human Services a part of the Standards for Privacy of Individually Identifiable Health Information under 45 CFR Parts 160 and 164.
d. Why are you providing $15 for people who choose to withdraw from the study? Isn’t this an incentive to withdraw?
This was an error that has been corrected in the current version of the consent form. We will pay $10 per completed study visit (i.e., the initial enrollment visit and each of the two telephone visits) for a possible total of $30 per patient. The payments will be made for each visit completed regardless of whether the subjects complete the study, so as not to coerce subjects into completing the study.
e. The consent form says that SSNs may be disclosed to the business office. Please provide a justification for collecting SSNs, including alternatives you considered and why they were not feasible.
Social security numbers are required by the UIC business office for tax reporting purposes when a sum $600 or more is payed to consultants or study subjects. Since the amount we are paying is less than $600, we have removed this language from the consent form and will not be collecting SSNs.
| File Type | application/msword |
| Author | Daniel Touchette |
| Last Modified By | Daniel Touchette |
| File Modified | 2007-08-22 |
| File Created | 2007-08-22 |
© 2026 OMB.report | Privacy Policy